基于苝核心的A-D-A型有机太阳能电池受体分子的理论研究*
2022-11-24秦银彬郑惠文汪易慧芝金宏璋肖思国
秦银彬,郑惠文,汪易慧芝,金宏璋,徐 炎,肖思国,闫 磊
(湘潭大学 物理与光电工程学院,湖南 湘潭 411105)
0 引言
太阳能是可持续未来的关键组成部分.随着能源危机与环境污染的日益严重, 如何利用清洁无污染的方法获得电能已成为当今社会的重要议题.与煤油、石油、天然气等不可再生、储量有限且对环境污染严重的化石燃料不同,太阳能取之不尽,用之不竭,绿色环保,是理想的清洁能源.有机太阳能电池的出现使人类获得了一种利用太阳能产生电能的方法.有机太阳能采用有机光敏材料作为吸光层,利用光伏效应产生电压形成电流,实现太阳能发电.与无机太阳能电池相比,有机太阳能电池具有独特的优势:有机材料的分子结构可以自行设计,通过化学合成制得,材料来源十分广泛;有机材料的光吸收系数高,可以降低电池厚度,减轻电池重量;有机太阳能电池可采用简单的真空蒸发和涂层工艺制模,对设备要求低,成本低;有机太阳能电池可制备在可弯曲折叠的柔性衬底上,利用卷对卷工艺可实现大规模制备[1-2].目前,交互型体异质结有机太阳能电池有较高的光电转换效率,其光电转换效率超过了18%[3],有很好的应用前景.有机太阳能的关键部分是活化层,活化层由电子给体和电子受体共混制备而成,给体材料一般由给体单元和受体单元共聚形成的高分子聚合物(D-A型或A-D-A型)组成.富勒烯及其衍生物和聚合物给体材料混合制成的电池器件光电转换效率已经超过 10%[4-5],然而富勒烯受体材料也存在着很多缺点,限制了太阳能电池器件光电转换效率的进一步提高,如可见光及近红外区对太阳光的吸收非常弱,LUMO能级不易调节,限制了太阳能器件开路电压的提高,化学合成复杂,成本高等[6].与PCBM相比,非富勒烯及其衍生物的受体材料具有可调节的LUMO能级,分子结构简单易合成以及可见光区域有更强且宽的吸收等优点,并且非富勒烯为受体的有机太阳能电池的光电转换效率已经超过了19%[5, 7-10].
大量研究表明,苝类材料是一类特殊的稠环结构化合物,苝二酰亚胺类衍生物是研究最早最广泛的有机太阳能电池半导体材料之一,由于它们的吸光特性好,太阳光谱响应范围宽,具有共轭平面结构,强拉电子的酰胺基团使其有高的电子亲和力和电子迁移率,是一种典型的N型电子受体材料[11].苝衍生物作为受体材料的研究还鲜有报道,因此,本文设计了两种以苝单元为核,以2-(3-氰基亚甲基)靛酮(INCN)作为受体单元的A-D-A型有机太阳能电池受体分子A1和A2,其中烷基链的引入是为了改善分子的溶解性及光物理性质.INCN作为强吸电子单元,可以促进分子内电荷的有效分离,增宽光的吸收范围.这种具有稠环结构的A-D-A型分子是目前最有效的分子构型之一,它们具有共同的结构特征:强吸电子单元分布于分子两端,并在固态堆积时,会发挥其吸电子的作用,促进分子间的电荷传输;分子有较好的平面性,可以促进分子内的电荷传输和分子堆积,有利于提高电荷的迁移率[12].氰基取代基具有强缺电子性质,在π共轭体系中能够有效地降低能级,因此被广泛应用于 N型半导体受体材料的设计中.目前高效的稠环电子受体几乎都含氰基官能团,氰基不仅能够大幅降低π共轭体系的能级,还在促进激子解离等方面具有重要作用.本文用密度泛函理论(DFT)、含时密度泛函理论(TD-DFT)研究了两种受体分子A1和A2的电子性质、光学性质以及分子内电荷转移性质,并计算了它们的单点能、静电势(ESP)分布、前线分子轨道及能隙、电子亲和能(Electron Affinity,EA)、吸收光谱、振子强度等,分析比较了两种分子作为有机太阳能受体分子的可行性,为实验筛选新的受体材料提供了新的理论依据.
1 计算方法
侧链烷基链对有机分子的计算结果几乎没有影响[13],为了节约计算时间和成本,本文用甲基取代了N原子处连接的烷基链,所设计的两种受体分子A1和A2的结构式如图1所示.

图1 两种受体分子的结构式Fig.1 The chemical structure of the two acceptors
1.1 分子的构象搜索
众所周知,构象是描述分子结构性质最广泛使用的概念.它是指具有一定构型的分子由于单键旋转而具有的空间结构性质.理论上讲,由于单键的旋转所导致的相同构型分子的构象是无数的,但是由于原子和基团间的相互作用,同一分子在自然条件下常常保持一种或几种热力学稳定的构象[13].本文通过Molclus程序[14]、 MOPAC程序[15]以及Gaussian09程序[16],对两种受体分子A1和A2进行了分子构象的搜索.如图1所示,受体分子A1和A2分别有两个可以旋转的单键1和2.设每个键每次旋转60°,那么总共就可以产生6×6=36个初始构象,用MOPAC程序[15]的PM6-D3半经验方法在LBFGS优化模式下对这些构象进行优化,得到一些能量极小的结构,再用优化精度更高的Gaussian09程序对MOPAC优化出的结构在DFT/B3LYP/6-31G(d, p)水平上进行再一次优化,筛选出能量最小的结构.对两种受体分子A1和A2用上述方法进行构象搜索,均只得到一种最稳定的构象结构,如图2所示.
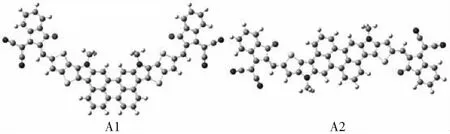
图2 用半经验方法PM6-D3和DFT/B3LYP/6-31G (d, p) 筛选后的分子构象Fig.2 The molecular conformation screened by semi-empirical methods PM6-D3 and DFT/B3LYP/ 6-31G (d, p)
由图2可知,A1和A2均只有一个构象,具有较好的平面结构,一方面可能是因为苝本身是一种稠环结构,具有很好的π共轭;另一方面可能是因为S-O键之间产生了氢键这种非共价相互作用,从而达到了“构象锁”的效果[17].
1.2 计算过程
DFT是模拟有机化合物电子结构和光学性质的一种合理方法[18-19].本文在DFT/B3LYP/GEN(H原子采用6-31G(d, p)基组,对C、N、O、S原子采用6-311G(d, p)基组)计算水平上对A1和A2进行了结构优化和频率计算.计算化学的理论方法定义了如何去近似求解分子体系薛定谔方程,而基组则决定了某种理论方法在实际计算过程中能达到的数值求解精度.交换-相关泛函B3LYP在1994年被提出后,几乎成了计算各种体系问题的默认方法.由于泛函B3LYP对弱相互作用力的计算存在误差,所以在计算过程中,加上了BJ阻尼的D3矫正,使计算结果更加可靠,以下涉及B3LYP的计算均加了BJ阻尼的D3矫正.
基于上述泛函和基组优化后的结构,本文分别在TD-DFT/CAM-B3LYP/6-311G(d, p)和DFT/M06-2X/DEF2TZVP水平上计算了分子A1和A2的激发态、吸收光谱等光学性质和单点能.为了能更好地分析两个分子中的电荷转移情况,基于Marcus理论[20]计算了两个受体分子A1和A2的内重组能λe.以上优化过程得到的分子结构均没有产生虚频,表明经过优化获得的几何结构是稳定的.
2 结果与讨论
分别用DFT和TD-DFT分析了受体分子的光学性质以及电荷传输性质等,具体结果如下.
2.1 前线分子轨道
前线分子轨道理论,是一种分子轨道理论,这一理论将分子周围分布的电子云根据能量细分为不同能级的分子轨道:有电子排布的,被称为能量最高的分子轨道HOMO;没被电子占据的,被称为能量最低的分子轨道LUMO.HOMO能级和LUMO能级是一个体系发生化学反应的关键,这两个轨道决定了分子的电子得失和转移能力以及分子间反应的空间取向等重要化学性质.图3和图4分别展示了A1和A2分子在DFT/B3LYP/GEN(H原子采用6-31G(d, p)基组,C、N、O、S原子采用6-311G(d, p)基组)模拟计算得到的前线分子轨道图.
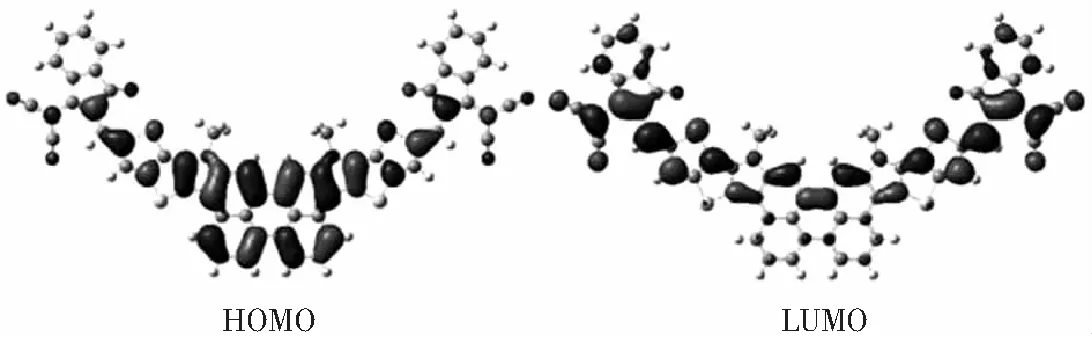
图3 A1分子在DFT/B3LYP/GEN计算水平下的能级轨道分布图Fig.3 Energy level orbital distribution diagram of A1 at DFT/B3LYP/GEN calculation level
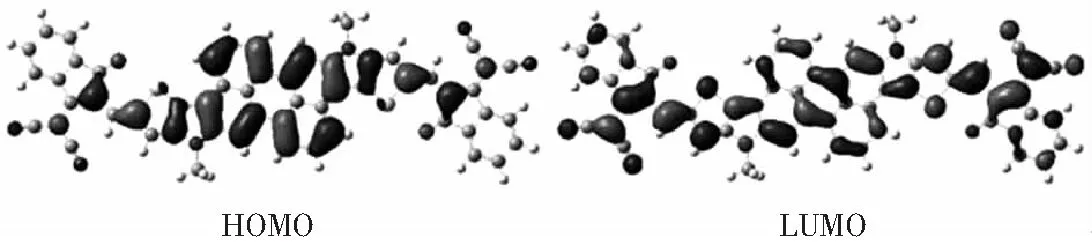
图4 A2分子在DFT/B3LYP/GEN计算水平下的能级轨道分布图Fig.4 Energy level orbital distribution diagram of A2 at DFT/B3LYP/GEN calculation level
从图3和图4中可以看出,A1和A2分子的HOMO能级的电子云主要分布在苝单元上.A1分子中的LUMO能级电子云主要分布在分子两端2-(3-氰基亚甲基)上,A2分子中的LUMO能级电子云分布在整个分子上.与A2相比,A1分子的HOMO能级和LUMO能级之间交叠部分较少,表明A1分子有明显的电子转移.激发过程中,电子从HOMO电子云分布区域向LUMO电子云分布区域转移[21].
2.2 分子的HOMO和LUMO带隙
分子体系的带隙有三种类型,分别是HOMO-LUMO带隙,基础带隙,光学带隙.其中,基础带隙是垂直电离能(Vertical Ionization Potential,VIP)与垂直电子亲和能(Vertical Electron Affinity,VEA)的差,光学带隙是指基态电子态通过吸收光子所能跃迁到的最低激发态对应的激发能.HOMO-LUMO带隙不能被实验方法测定.本文用DFT/B3LYP/GEN(混合基组,H原子采用6-31G(d, p)基组,C、N、O、S原子采用6-311G(d, p)基组)模拟计算了两个受体分子A1和A2在真空条件下的HOMO-LUMO带隙,结果如图5所示.
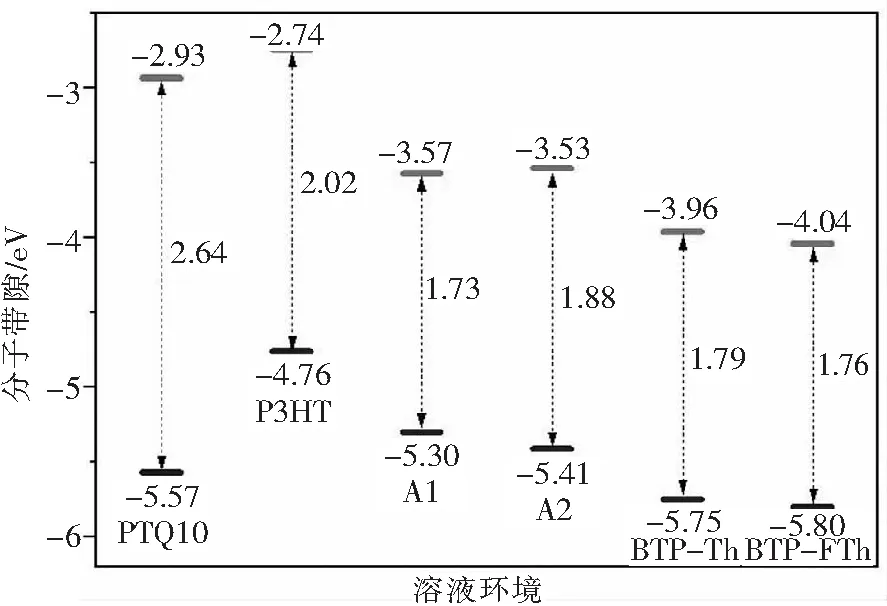
图5 PTQ10、P3HT给体和A1、A2以及BTP-Th和BTP-FTh受体的HOMO-LUMO能级图Fig.5 HOMO-LUMO level diagrams of PTQ10, P3HT donor and A1, A2,BTP-Th and BTP-FTh acceptors
以PTQ10为给体,BTP-Th和BTP-FTh为受体的有机太阳能电池效率已经超过了19%[10],如图5所示,A1和A2的HOMO-LUMO能级与给体P3HT能很好地匹配,且和给体材料吸收不同的太阳光谱段,没有产生光竞争吸收.A1分子的带隙值与目前效率最高的非富勒烯受体分子BTP-Th和BTP-FTh的带隙值比较接近.A1和A2分子在DFT/B3LYP/GEN计算水平下的真空和溶剂中的能隙如表1所示.用隐式溶剂模型(SMD)考虑了二氯甲烷溶剂效应.表1表明,A1和A2分子在真空或溶剂条件下都有较低的LUMO能级,有利于从给体材料上接收电子.

表1 A1和A2分子在DFT/B3LYP/GEN计算水平下的真空和溶剂中的能隙
2.3 UV-Vis吸收光谱
分子的吸收光谱可以反映分子对光的吸收能力,优质的有机太阳能电池受体分子的光吸收范围应该和给体分子互补,两者有比较少的光竞争吸收.本文用TD-DFT/CAM-B3LYP/6-311G(d, p)计算模拟了两个分子A1和A2分别在真空和溶剂条件下的吸收光谱.采用TD-DFT计算了30个激发态.在TD-DFT计算中,用隐式溶剂模型(SMD)考虑了二氯甲烷溶剂效应.如图6所示,在相同条件下,与A2的吸收峰相比,A1发生了红移,符合它们的能隙特征,A1的能隙更窄.与真空环境相比,A1或A2分子在溶剂环境中的吸收峰,也发生了红移.可能是因为模拟的溶剂是二氯甲烷,是一种极性溶剂.溶剂分子和溶质分子会存在多种作用力.在极性溶剂中,溶质分子的极性基团会受偶极作用、静电作用以及氢键等影响,基团振动更加明显,其键力常数减小而发生吸收峰的红移现象.在真空环境中,A1和A2的吸收峰分别为616.73 nm、591.33 nm,振子强度分别为2.842 3、3.1912;在溶剂环境下,A1和A2的吸收峰分别为649.98 nm、628.3 nm, 振子强度分别为3.078 1、3.547 8.
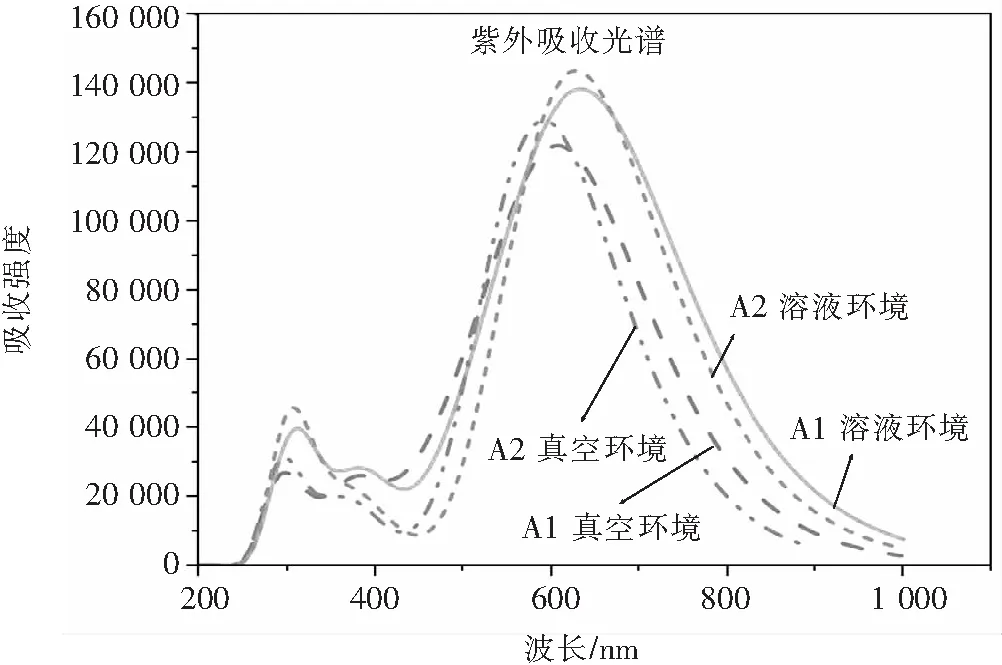
图6 A1和A2分子在TD-DFT/CAM-B3LYP/6-311G(d, p)计算水平下的真空和溶液中的紫外吸收光谱Fig.6 UV absorption spectra of A1 and A2 in vacuum and solution at TD-DFT/CAM-B3LYP/ 6-311g (d, P) calculation level
2.4 静电势
通过静电势可以预测分子的一些性质,静电势的含义是把一个正电荷从无穷远处拉到图上某个位置所需要做的功的大小.用0.001 a.u.的等值面作静电势图.由图7的静电势分布图可知,A1和A2分子中,蓝色区域主要集中在与苝相连的甲基部分,有利于吸引负电荷,红色区域主要集中在(-CN)氰基和氧原子附近,是因为氧原子和氰基上的N原子含有孤对电子.同时受体分子和给体分子的静电势分布不同在共混时会形成分子间的电场,从而促进激子解离,促进电子从给体转移到受体.

图7 在DFT/B3LYP/GEN水平下计算的分子静电势图Fig.7 Molecular electrostatic potential diagram calculated at DFT/B3LYP/GEN level
2.5 电子激发分析
用TD-DFT的方法,在CAM-B3LYP以及基组6-311G(d, p)条件下对分子A1和A2进行激发态计算,用Multiwfn[22]程序对计算结果做了电子激发分析,分析两个分子的电子激发过程.A1和A2分子的前五个激发态的数据如表2和表3所示.
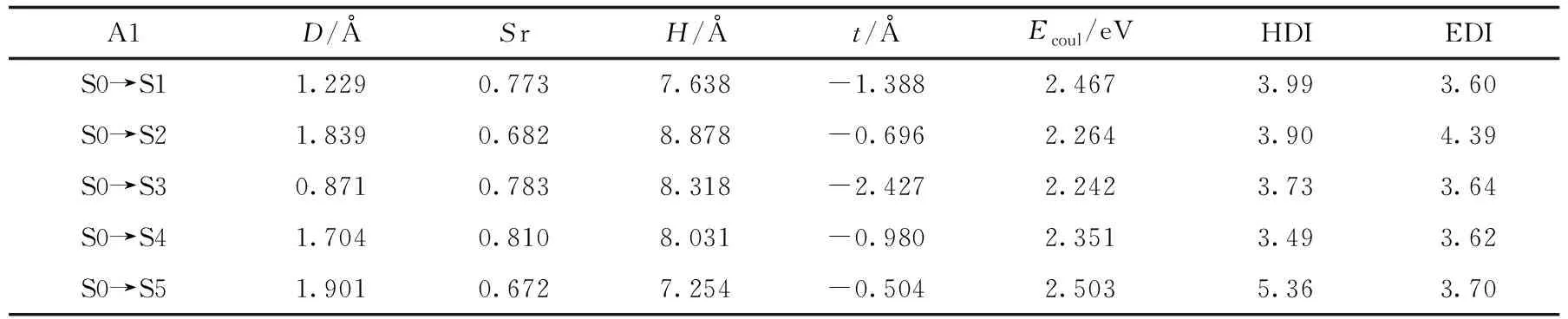
表2 A1分子在TD-DFT/CAM-B3LYP/6-311G(d, p)的电子激发数据
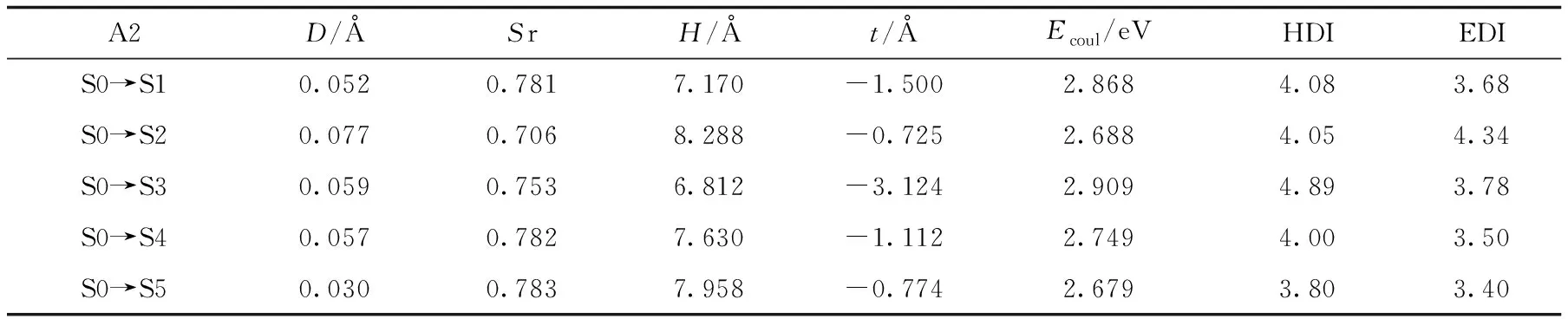
表3 A2分子在TD-DFT/CAM-B3LYP/6-311G(d, p)下的电子激发数据
在表2和表3中,D指数定义了衡量空穴和电子质心之间距离,D指数由以下公式定义:
DX=|Xele-Xhole|,
DY=|Yele-Yhole|,
DZ=|Zele-Zhole|,
(1)
式(1)中:Xhole、Xele分别为空穴和电子质心的x坐标;Yhole、Yele分别为空穴和电子质心的y坐标;Zhole、Zele分别为空穴和电子质心的z坐标.Xhole、Xele分别由ρhole和ρele函数乘上x坐标变量在全空间积分得到,Yhole、Yele和Zhole、Zele类似.t指数衡量空穴和电子的分离程度,t=D-HCT,t>0表示电子和空穴分离较为充分.其中,HCT=|H·uCT|,uCT是指电荷转移方向上的单位矢量,H是Hx、Hy、Hz合在一起写成的矢量,Hx、Hy、Hz是H分别在x,y,z方向上的投影.Hλ=(σele,λ+σhole,λ)/2,λ={x,y,z},Hindex=(|σele|+|σhole|)/2.其中,σ的三个分量x,y,z相当于空穴或者电子在x,y,z方向上分布的方均根偏差,体现了空穴和电子的分布广度和弥散程度.
σ对于空穴的x分量定义为:
(2)
σ的模,即x,y,z三个分量的平方和开根号,衡量空穴和电子的整体分布广度.
H指数可以用来表征电子和空穴的平均分布情况,H指数越大,表示电子和空穴的平均分布广度越广.Sr指数表示电子和空穴的重合程度:
(3)
式(3)中:r表示坐标矢量;ρhole和ρele分别代表空穴和电子的分布函数;Sr数值越小代表电子和空穴重叠程度越小.空穴离域指数(hole delocalization index, HDI)和电子离域指数(electron delocalization index, EDI)表明了电子的空穴离域程度,HDI(EDI)数值越小,说明空穴(电子)离域程度越高.空穴-电子库仑吸引能Ecoul(eV)和电子激发特征有密切关联,对其影响最大的是D.D越大,空穴和电子主体分布越远,因此空穴和电子的库仑相互作用应当更弱[23].上表中A1的各个激发态D指数均大于A2的激发态D指数,说明A1分子的电子传输距离比A2分子的传输距离远,t指数的值也说明了这一点.A1分子激发态t指数更接近0.由于A1分子各个激发态的D指数比A2分子的大,分子中的空穴和电子主体分布越远,因此空穴和电子的库仑相互作用更弱.空穴和电子的库伦吸引力越弱,越容易在光的激发下分离.使用Multiwfn[22]程序对分子做电子激发分析和查看所有激发态中的主要轨道跃迁贡献来区分分子A1和A2分子的电子激发类型,轨道贡献见表4.
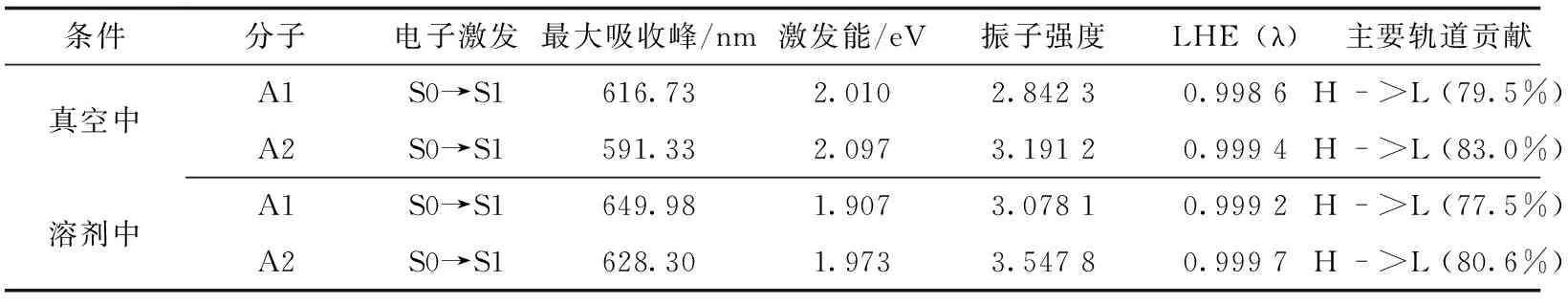
表4 在TD-DFT/CAM-B3LYP/6-311G(d, p)水平下对A1和A2的激发态分析
LHE是光捕获效率,LHE(λ)的值可以通过以下公式求得[24-26]:
LHE(λ)=1-10-f,
(4)
式(4)中:f表示波峰处的振子强度.由于跃迁过程对电子自旋状态的限制,一般产生的是单线态激子.所以在太阳能电池中,单线态激子具有重要作用[12].从表4可以看出,A1和A2在真空条件和溶剂条件下的最大吸收峰对应其第一单线态激发态.由于电子从HOMO能级跃迁到LUMO能级的轨道贡献率都大于75%,占绝对主导性,比如大于75%,所以通过分析这两个轨道的特征来判断激发的类型[21].HOMO和LUMO的平面图见图3和图4.HOMO能级和LUMO能级的截面图见图8.
从图8可以看出,A1和A2的电子从基态跃迁到单线态第一激发态的激发属于成键轨道π到反键轨道π*的激发.结合表2和表3 的数据和图3图4来看,A1分子的激发属于电荷转移激发,A2属于局部激发.
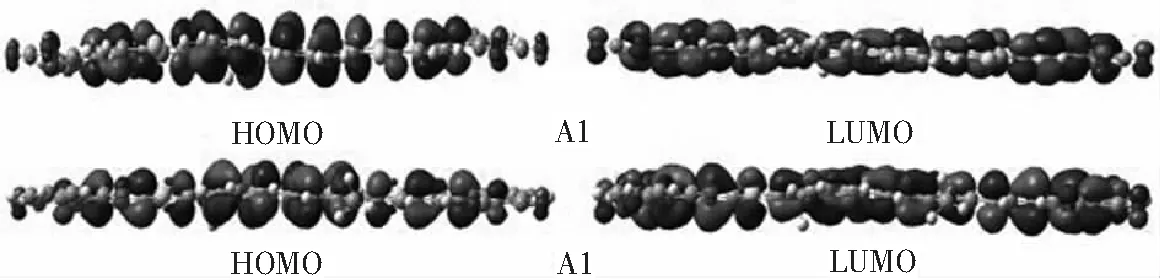
图8 A1和A2在DFT/B3LYP/GEN计算水平下的前线分子轨道截面图Fig.8 A1 and A2 forward molecular orbit cross sections at DFT/B3LYP/GEN computing level
为了进一步阐明电子转移的特性,进行了激发态与基态之间的电子密度差(Charge Density Difference, CDD)分析[27],并在0.000 5 a.u.的等值面下绘制了两个分子第一激发态单线态的电子密度差等值面图.绿色区域代表电子密度增加的地方,蓝色区域代表电子减少的地方.由图9可知,在A1和A2分子内电荷的转移方向为从苝给体单元向两端的2-(3-氰基亚甲基)靛酮(INCN)受体单元转移.
图9 在TD-DFT/CAM-B3LYP/6-311G(d, p)下的电子密度差CDD图Fig.9 CDD diagram of electron density difference at TD-DFT/ CAM-B3LYp / 6-311G (d, p)
为了进一步了解电子转移情况,如图10分块方式把A1和A2分子从左至右分成5个片段,用Multiwfn程序计算电子激发过程中任意片段间的电子转移量.计算结果如表5所示.
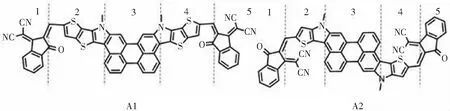
图10 A1和A2的分块情况Fig.10 The block way of A1 and A2
负值表示电荷量减少,正值表示电荷量增加,从表5可以看出,A1和A2的电子转移方向和图9的CDD图相符.
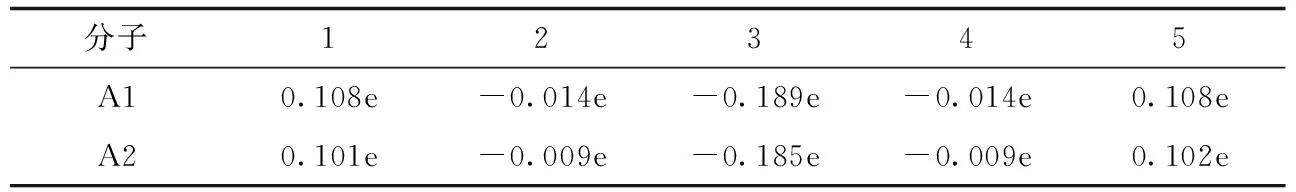
表5 在TD-DFT/CAM-B3LYP/6-311G(d, p)水平下的电荷转移量
2.6 偶极矩、电子亲和能
分子内偶极矩对保持体系的电荷转移特性起着非常重要的作用[28-29],偶极矩与空穴-电子对的分离难易程度有关,偶极矩越大,则空穴-电子对越容易分离,同时偶极矩越大,光伏性能越好[30].从表6可以看出,A1的偶极矩大概是A2的9倍.分别计算了A1和A2分子的绝热电子亲和能,即用DFT/B3LYP/6-31G(d, p)优化中性分子后的单点能减去用DFT/B3LYP/6-31G(d, p)优化阴离子结构后的阴离子单点能[31].单点能在DFT/M06-2X/DEF2TZVP水平下计算,同时加上了零阻尼的D3矫正,计算阴离子单点能在DEF2TZVP基组上加了弥散函数.电子亲和能表征了分子吸引电子的能力,作为电子受体分子来说,好的受体分子的电子亲和能越高.从表6中可以看出,A1分子的电子亲和能更大,表明A1分子的吸电子能力大于A2分子.

表6 DFT/M06-2X/DEF2TZVP下计算的电子亲和能和DFT/B3LYP/GEN下计算的偶极矩
2.7 内重组能和电荷迁移速率
外部(ext)和内部(int)重组能是两种重组能.具体分为外重组能[32]和内重组能,前者考虑了外部环境中的弛豫效应和电荷输运过程中的极化效应,而后者考虑了分子内部几何结构的快速变化.溶液环境下往往经验性地取0.2~0.6 eV作为外重组的值,本文采用M06-2X泛函和DEF2TZVP基组计算了两种分子的内重组能,M06-2X泛函加上了D3零阻尼矫正使结果更准确.内重组能由以下公式得到[31]:
(5)
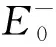
电子迁移数据可由以下式子得到[33]:
(6)
式(6)中:λe为式(5)中的内重组能;V为电子耦合强度,由Koopman定理得,电子耦合强度为中性体系LUMO+1与LUMO能量差的一半[34];ћ为普朗克常数;KB是玻尔兹曼常数;T代表温度,本文取300 K.最终计算结果见表,从表7中可以看到,A1分子和A2分子都有高的电荷迁移率.太阳能电池的受体分子需要有高的电子迁移率,并且最好与给体材料的空穴迁移率相平衡,以减少能量损失,保证高的电荷载流子传输效率和高的填充因子.

表7 A1和A2分子的重组能和电荷迁移率
3 结论
本文利用密度泛函理论方法, 结合 Marcus Hush电荷传输模型, 理论研究了A1和A2的分子结构、电子性质、光吸收性质,结果表明:(1)A1受体分子具有低的HOMO 能级, 较窄的光学带隙, 在紫外-可见区具有强烈的光学吸收;(2)A1和A2分子均有较高的电荷迁移率;(3)A1具有对称结构有利于提高结晶性,A2具有不对称的结构有利于降低结晶性,可以有效调控与高分子给体体系的共混时的形貌,使其在纳米尺度上达到小尺寸的相分离,从而提高有机太阳能电池器件的性能.A1分子和A2分子是一种非常有发展前景的有机太阳能电池电子受体材料,值得实验上进一步合成及器件优化研究.
