高端制剂的临床可及性研究
2022-11-04由春娜
由春娜
烟台大学药学院
邵亚婷
绿叶制药集团有限公司
董敏
烟台大学药学院
傅风华*
烟台大学药学院
周建平*
中国药科大学
李大魁*
北京协和医院药剂科
1 高端制剂开发现状
2016年,工业和信息化部出台了《医药工业发展规划指南》[1],指出要重点发展高端制剂,包括脂质体、脂微球、纳米制剂等新型注射给药系统,口服速释、缓控释、多颗粒系统等口服调释给药系统,经皮和黏膜给药系统,儿童等特殊人群适用剂型等,推动高端制剂达到国际先进质量标准。上述关于高端制剂的范围,为工业和信息化部所划定。我国目前药品监管相关法规、指南中,尚无明确的定义及范围。在不同的药品监管法规和指南中,多以特殊制剂或特殊剂型泛指这类制剂,且名称和范围有所不同。
(1)2007年版《药品注册管理办法》[2]将靶向制剂、缓释制剂、控释制剂等定义为特殊剂型。
(2)在《化学药物制剂人体生物利用度和生物等效性研究技术指导原则》[3]中,特殊制剂的范围包括口服缓(控)释制剂、特殊活性成分制剂、复方制剂。
(3)在《已有国家标准化学药品研究技术指导原则》[4]中,特殊注射剂的范围包括脂质体、乳剂、微囊(球)等,其质量和活性成分的体内行为受处方和工艺的影响较大,引起药物在体内分布和消除的差异。
(4)《化学药品注射剂(特殊注射剂)仿制药质量和疗效一致性评价技术要求》[5]指出:“特殊注射剂是指与普通注射剂相比,特殊注射剂的质量及其活性成分的体内行为受处方和工艺的影响较大,可能进一步影响制剂在体内的安全性和有效性,例如脂质体、静脉乳、微球、混悬型注射剂、油溶液、胶束等”。
类似于工业和信息化部提出的高端制剂,美国食品药品监督管理局(FDA)是在其发布的《仿制药使用者付费法案》(GDUFA)Ⅱ承诺书(GDUFAⅡCommitment Letter)中界定了复杂制剂的范围,包括[6]:①含有复杂的活性成分(如肽类、高分子化合物、化学原料药的复杂混合物、天然来源成分)。②复杂的制剂处方(如脂质体、胶体)。③复杂的递送途径(如局部作用产品、皮肤病产品、复合眼科产品,配制为悬浮液、乳液或凝胶耳用剂型)。④复杂的剂型(如透皮贴剂、吸入剂、缓释注射剂)。⑤复杂的药械组合产品(如自动注射剂、计量吸入剂)。⑥其他产品,其批准途径或可能的替代方法的复杂性可能不确定,早期与监管机构开展科学性的沟通会使其受益。美国FDA 定义的复杂制剂与我国工业和信息化部提出的高端制剂,所涵盖的范围基本相近,但FDA规定的复杂产品还涵盖了复杂活性成分的产品,这类产品不在本文讨论范围之内。
高端制剂对于某些严重疾病的治疗至关重要,并具有普通制剂所不具备的临床优势。例如:①长效给药:用于治疗精神分裂症的棕榈酸帕利哌酮开发成1 个月或3 个月甚至6 个月给药一次的长效注射剂,提高了顺应性的同时也减少了复发率。②提高药物的安全性:将紫杉醇开发成脂质体,避免了紫杉醇注射剂中的聚氧乙烯(35)蓖麻油引起的不良反应。盐酸多柔比星脂质体与普通注射液相比,可降低不良反应发生率。透皮贴剂作为一种经皮给药系统,药物以一定速率透过皮肤经毛细血管吸收进入体循环,可以避免肝脏的首过效应和胃肠因素的干扰,避免药物对胃肠道的不良反应,长时间维持恒定的血药浓度,避免峰谷现象,特别适用于老年人及不宜口服给药的患者。③病灶部位定位给药:吸入制剂通过口鼻给药,可将药物直接递送至呼吸系统用于治疗呼吸疾病,能够实现药物的定位给药,是治疗呼吸系统疾病最理想的给药方式,同时吸入制剂可以避免胃肠道给药产生的首过效应,提高生物利用度。
基于上述临床优势,高端制剂具有很高的市场效应(表1)。近些年上市的部分高端制剂,上市后市场份额持续增长,其产生的市场效应不亚于创新药。但是部分高端制剂由于技术壁垒高、仿制难度大,长期没有仿制药上市(表2),竞争不充分。以表1 中的亮丙瑞林微球为例,原研药已上市30 余年,每年仍有6 亿~10 亿美元的销售额,但是目前我国除原研药外,只有2 家早年间获批的仿制药,这2家仿制药目前均未通过一致性评价。

表1 部分高端制剂的全球销售额
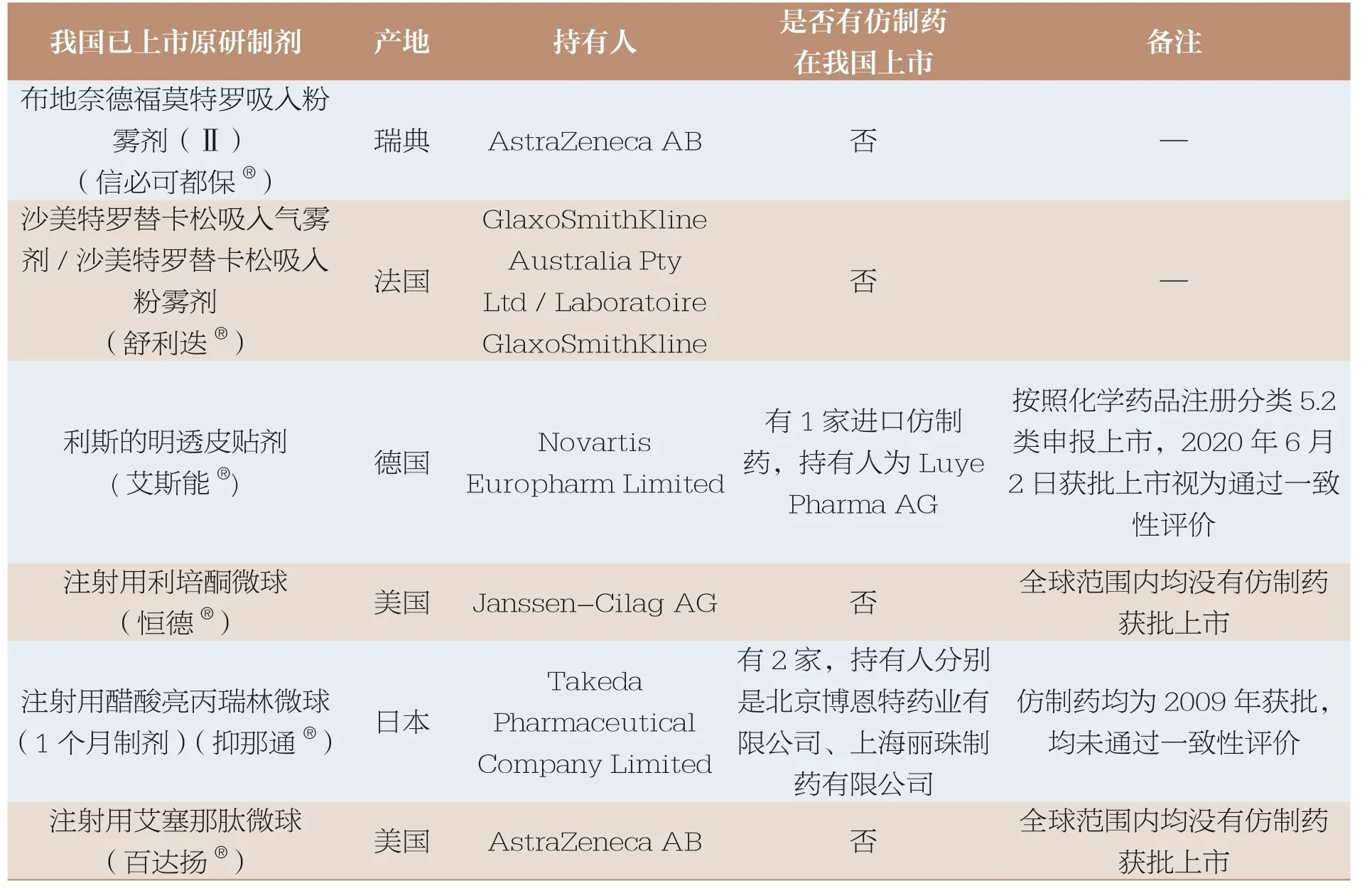
表2 部分高端制剂仿制药上市情况
2 高端制剂开发面临的挑战
2.1 关键技术/辅料/设备被垄断
高端制剂的研发不同于传统制剂,其技术壁垒高,如复杂的制备工艺、生产设备、功能性辅料以及表征手段等,导致高端制剂的开发难度大。
2.1.1 关键技术被垄断
高端制剂的开发,具有很高的技术开发及规模化商业生产的难度,关键技术基本被几家企业垄断,这是高端制剂难以开发、仿制的根本原因之一。以注射用微球制剂为例,其给药技术被瑞士Debiopharm 公司、美国Alkermes 公司、日本武田公司等仅有的几家跨国公司掌握,并开发了相关产品。我国目前仅有少数几家公司掌握了部分长效缓释注射剂技术,如绿叶制药、石药集团、恒瑞医药、丽珠制药、科伦药业、齐鲁制药等。
2.1.2 关键辅料市场处于垄断状态
高端制剂中的功能性辅料是其核心之一,也是高端制剂实现其药物递送的载体。在高端制剂用功能性辅料领域,大多被生产技术较成熟的国外大型化工企业垄断,主要供应商包括陶氏、杜邦、巴斯夫、瓦克化学、亚什兰、阿科玛、赢创等[7],美国陶氏化学垄断中国聚乙二醇类[8]。以生产透皮贴片的关键辅料压敏胶为例,目前登记平台收载的11 家压敏胶,有10 家是境外企业,形成垄断之势(表3)。
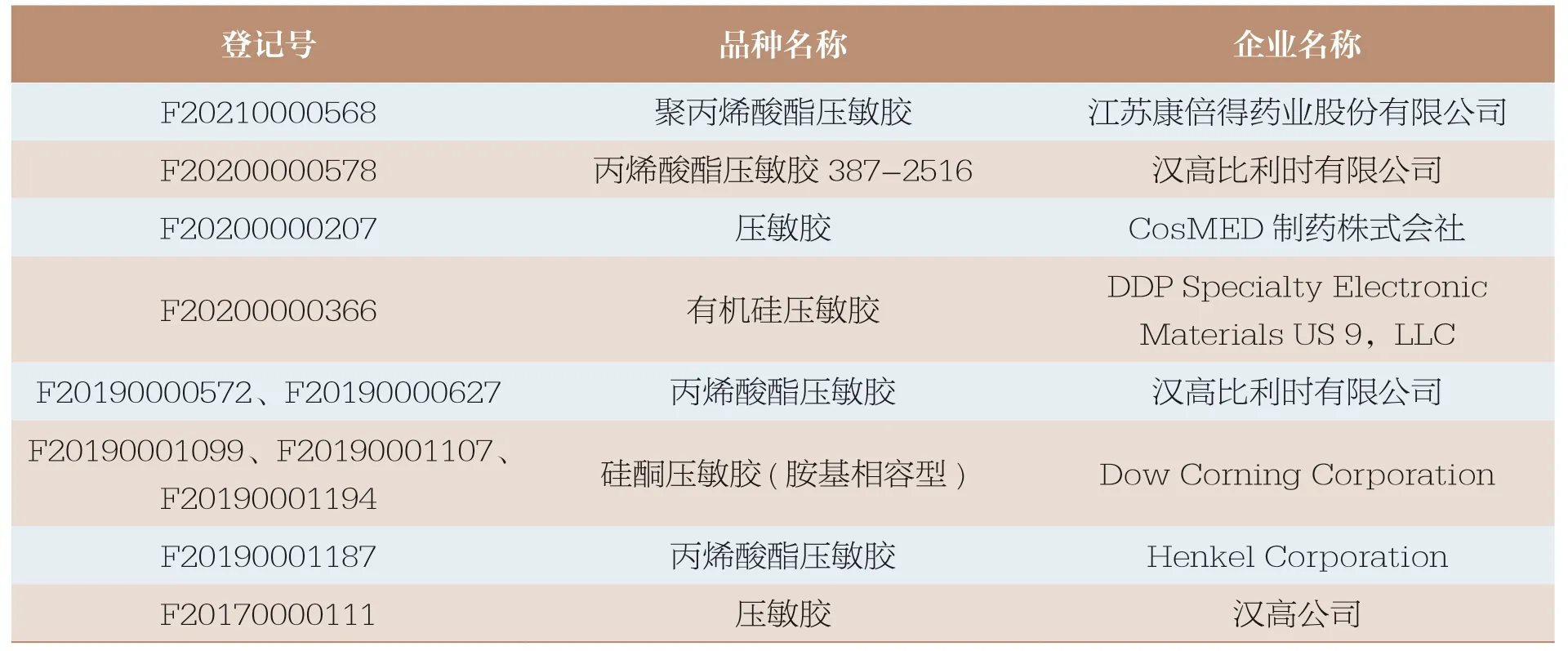
表3 原辅包登记平台收载的压敏胶
功能性辅料被国外企业垄断的现状导致高端制剂开发的成本高昂,技术难度大幅增加。以《化学药品注射剂(特殊注射剂)仿制药质量和疗效一致性评价技术要求》中提到的特殊注射剂为例,注射剂仿制药需满足Q1、Q2 和Q3 的要求,即辅料种类、辅料用量、制剂特性均与参比制剂相同。但我国特殊注射剂的功能性辅料大多依赖进口,如制备微球所用的可降解载体高分子材料聚乳酸(PLA)和丙交酯乙交酯共聚物(PLGA)价格昂贵,且部分微球原研制剂所用的PLA/PLGA 为原研药厂家定制,其他公司基本不可得。又如,聚乙二醇(PEG)化磷脂是纳米脂质制剂如脂质体注射剂、脂质纳米粒(mRNA、siRNA、pDNA 等核酸递送载体)必不可少的辅料。早在2016年,《医药工业发展规划指南》就已经提到需重点发展PEG 化磷脂、抗体修饰用磷脂等功能性合成磷脂等面向高端制剂的辅料,但目前国际上能够稳定供给PEG 化磷脂的企业数量并不多,处于垄断状态,其进口单价按克计算,价格居高不下,进一步加大了这类高端制剂的研发技术壁垒。
2.1.3 关键设备往往需要自行设计和定制
高端制剂生产所用的关键设备往往需要自行设计和定制,在设计期间,可供参考用的专利、案例等非常少,与一般的口服制剂、注射剂相比,高端制剂的生产线建设,设计复杂、周期长且费用高昂,无疑又加剧了这类产品的开发难度。以透皮贴片为例,涂布、干燥、裁切等关键工艺所用的设备,均需要根据品种特点,单独设计、定制。
2.2 高端制剂处方开发难度大
高端制剂体内释药机制十分复杂,尤其是一些聚合物胶束、脂质体等纳米颗粒结构的药物[9],难以建立体内外相关性,往往需要依赖动物或人体的药代动力学研究进行处方开发,进一步加剧了处方开发难度。
以棕榈酸帕利哌酮注射液(Invega Sustenna®)为例[10],Janssen 在研制该药物期间,共设计了13 个处方,其中5 个处方开展了临床药理学,合计纳入732 例受试者,用于处方开发。处方F11 用于Ⅱ期、Ⅲ期临床试验,后期在F11 的基础上又进一步进行了优化,最终得到了处方F13,用于申报上市。通过人体药代动力学研究进行处方开发费用十分高昂、周期长,开发耗费10年之久。
又如,诺华制药的利斯的明透皮贴剂(艾斯能®)[11],在开发早期,设计了5 个处方,经过临床试验获得药代动力学参数、黏附力、局部刺激性参数,确定目标处方。这与一般的口服制剂或注射剂相比,开发的难度、周期和费用均大幅增加。
除了需要通过临床试验来筛选高端制剂处方带来的开发难度,部分高端制剂从中试车间到商业化车间的生产规模放大技术难度也很大。在生产规模放大过程中,任何微小的变更都可能产生影响药物体内释药行为的风险。因此,需要更多批次的生产用于批量放大研究、工艺参数的调整等,较普通片剂、胶囊剂和注射液的处方工艺开发成本高。
2.3 高端制剂的临床开发周期长、投入高
一些高端制剂品种如植入剂、注射用微球、微晶制剂等具有长效释药的特点,在临床试验过程中,受试者给药间隔较一般制剂长,每次给药间隔1 周、1 个月,甚至3个月、6 个月,导致其临床试验周期长、入组受试者例数多,临床试验成本高昂。如针对治疗精神分裂症的储库型制剂,欧盟要求研究时应考虑以下几点[12]:①通过药代动力学临床研究结合其释药特性,来证明该制剂为储库制剂。②比较储库制剂与口服制剂的生物利用度,以评价活性成分可接受水平的持续时间。③在病情稳定的患者身上比较储库制剂与口服制剂的有效性。④解决从口服制剂到储库制剂的转换问题。⑤评估特定储库制剂的安全性。⑥通常需要通过临床试验比较口服制剂和储库制剂的有效性,以确定储库制剂给药间隔的合理性。依据上述要求,大冢制药在开发阿立哌唑长效肌内注射剂时,除开展3 项临床药理试验和群体药代动力学模拟分析之外,还开展了7项Ⅲ期临床试验,Ⅲ期临床研究包括1 项有效性研究、2 项阳性对照实验以及4 项大型开放性安全试验[13]。针对储库制剂释药特点的临床研究较普通剂型要求多且设计复杂,增大了其临床开发的难度。
2.4 高端制剂仿制难度大、挑战高
高端制剂难以仿制,是业内公认的事实。在仿制过程中面临着药学一致和生物等效的挑战。FDA 专门总结了复杂制剂难以仿制的几大因素[14]:复杂的处方和辅料、工艺和原料的微小变更可导致制剂的重大变更、制剂特性复杂、没有体外溶出标准、释药机制未被充分研究、几乎没有体内体外相关性(IVIVC)相关模型、复杂的生物等效性试验设计等。
以生物等效性试验设计为例,表4 为帕利哌酮缓释片和棕榈酸帕利哌酮注射液(1 个月制剂)的生物等效性试验对比表。试验设计、样本量、受试者、入组难度、脱落率、生物样品采样时长、费用等各方面,长效注射剂的生物等效性试验难度都远远高于口服缓释片。

表4 帕利哌酮缓释片和棕榈酸帕利哌酮注射液(1 个月制剂)生物等效性试验对比
对于高端制剂中的脂质体仿制药,生物等效性研究常常要求游离的和脂质体包封的药物的 AUC0-t、AUC0-∞、Cmax几何均值比值的90%置信区间数值应不低于80.00%,且不超过125.00%。这与普通口服制剂的生物等效性试验设计相比,难度增大。
部分高端制剂的欧美仿制药指南与我国临床实践不符,难以执行[15]。例如,棕榈酸帕利哌酮注射液,FDA 建议以使用该制剂稳定的患者作为受试者,由于原研药售价高昂,中国临床上使用该制剂的患者人数非常少,无法满足我国法规的试验要求。又如,FDA 关于阿立哌唑长效肌内注射剂的仿制药指南要求受试者为已使用阿立哌唑长效肌内注射剂且稳定的患者,但原研药未在中国上市,没有此类人群开展临床试验。
3 中美提升高端制剂开发的相关举措
3.1 美国复杂制剂相关鼓励举措
针对复杂制剂仿制药的开发,美国FDA 站在引导者的角度,集合多方面的力量,全面引导申请人开发复杂制剂仿制药。具体表现在以下几个方面。
3.1.1 明确规定复杂制剂的范畴
FDA 详细规定了复杂制剂的范畴,为进一步制定复杂制剂的相关政策指南,奠定了基础。
3.1.2 加速制定复杂制剂仿制药个药指南
FDA 专门成立了复杂制剂仿制药指南数据库,截至2021年12月30日,已经出台了超过400 个复杂制剂的仿制药指南。在GDUFA Ⅲ中,FDA 承诺从2023 财年开始,在批准后2年内对50%新获批的复杂制剂发布仿制药指南, 在批准后3年内对75%新获批的复杂制剂发布仿制药指南。其中,对于含有新的分子实体的复杂制剂新药,在批准后2年内对90%新复杂制剂发布仿制药指南[16]。
3.1.3 不断对已发布的复杂制剂仿制药指南进行完善
随着对复杂制剂研究的不断深入,FDA 对其理解愈加深刻、科学,进而基于科学研究结果修订仿制药个案指南。以两性霉素B 脂质体仿制药指南为例[17],自2014年4月发布首版指南之后,FDA 分别在2016年1月、2020年8月进行了两轮修订。与之前的版本相比,修订后的指南:①建议在健康受试者而非患者人群中进行生物等效性研究,以简化体内生物等效性研究设计和受试者招募。②关于游离(脂质体未结合)两性霉素 B 的数据可以用作支持证据,而不是建立生物等效性的关键证据。这样的修订提高了生物等效性研究的效率和灵活性,有助于仿制药的开发并改善临床试验。
3.1.4 发布复杂制剂行业指南
除个药指南之外,FDA 发布了超过30 份复杂制剂开发相关的行业指南,针对如透皮贴剂、脂质体、纳米给药系统、吸入制剂等复杂制剂的开发予以指导。
3.1.5 开通专门的复杂制剂沟通交流渠道
除出台相关技术指南外,在个性化指导申请人开发复杂制剂仿制药方面,FDA 开辟了多种沟通交流渠道。
针对仿制药, 申请人可提出受控通信(controlled correspondence)[18], 与FDA探讨一般的技术问题,申请人可通过受控通信与FDA 确认仿制药的处方设计等产品开发相关问题。这一沟通交流方式对复杂制剂仿制药开发尤为重要,以复杂注射剂为例,通常要求复杂注射剂仿制药的辅料种类和用量应与参比制剂相同(Q1/Q2),在参比制剂的处方组成和辅料用量无法获得的情况下,申请人通过文献查阅和对原研药进行拆方研究,初步确定仿制药的处方组成,可以通过受控通信与FDA 确认其目标处方设计是否符合Q1/Q2。FDA在确认支持性资料充分后,14个自然日通过邮件予以答复,就复杂注射剂处方予以确认,以提升复杂制剂仿制药开发成功的概率。
此外,FDA 还针对复杂制剂仿制药,专门开通了沟通交流通道:GDUFA 下复杂制剂ANDA申请人和FDA 之间的正式会议(Formal Meetings Between FDA and ANDA Applicants of Complex Products Under GDUFA)[19]。 申请人可在产品的早期开发阶段申请与FDA召开正式的沟通交流会议、药物研发沟通交流会(Product Development Meetings),以获得FDA 的开发指导。FDA 可对复杂制剂申请人的以下沟通作出回应:①临床内容的评价。②对参比制剂的生物等效性方案进行审查,其中包括风险评价和缓解策略,以及确保安全使用的要素。③要求对相同研究类型的替代生物等效性方法进行评价(如药代动力学、体外、临床)。此外,申请人还可以在产品正式递交上市申请前与FDA 召开上市申报前沟通交流会(Pre-Submission Meetings)。如果召开了上市申报前沟通交流会,那么在第一轮审评中期前,当FDA 发现重大问题时,会主动要求与申请人召开审评过程中的沟通交流会(Mid-Review-Cycle Meetings)。 这一系列举措旨在全面指导复杂制剂仿制药的开发。
3.1.6 资助复杂制剂相关研究课题、成立复杂制剂研究中心
为了能更快、更好地理解复杂制剂仿制药的特性,使其能够替代原研药,FDA 在GDUFA Ⅱ(2018-2022)中,每年安排15个复杂制剂的优先科学研究计划,并于2020年在马里兰大学和密歇根大学成立了复杂仿制药研究中心(CRCG)。FDA 联合学术界力求在复杂仿制药科学研究上取得突破,为进一步提升复杂制剂的公众用药可及性发挥其引导性作用。以资助课题为例,2020年之前资助的、2020年正在开展研究的课题有44 个,2020年新资助了10 个复杂制剂相关的课题,未来还将持续资助复杂制剂相关课题。
3.2 我国高端制剂相关指南
为了进一步扩大高端制剂的临床用药可及性,我国药品监管机构已经开始着手从各方面制定相关指南,包括行业指南和个药指南,加速推动高端制剂的开发(表5)。

表5 我国药品监管机构公布的高端制剂相关指南

续表
4 针对高端制剂开发的相关建议
为进一步提升我国高端制剂的开发,加快我国从制药大国向制药强国的转变,针对高端制剂上市前开发遇到的诸多难题,提出以下几点建议。
4.1 明确高端制剂的定义及范围
我国现行药品监管相关法规、指南未对高端制剂的范围进行界定。在不同的法规和指南中,名称和范围亦有所不同。建议药品监管机构针对高端制剂的特殊性,对其定义以及囊括的范围进行规定,为后续鼓励政策的顺利实施奠定基础。
4.2 进一步加快出台高端制剂仿制药指南
高端制剂的特殊性需要监管机构针对品种给予更多的指导,按品种制定仿制药指南,同时针对不同剂型,结合国家药品监督管理局药品审评中心已有的审评经验,制定行业指南,并随着科学积累的提升,不断地对已有的指南进行修订。建议:①加快制定微球、脂质体、混悬型注射剂等高端制剂的药学研究指南,指导高端制剂仿制药的开发。②加快制定微球、脂质体、混悬型注射剂等高端制剂仿制药生物等效性评价指南。③动态修订高端制剂开发的行业指南和高端制剂仿制药个药指南。
4.3 出台高端制剂研发的支持性政策
为了更好地鼓励和支持开发高端制剂,建议药品监管机构进一步重视高端制剂的发展,高度参与到高端制剂的开发过程中,给予行业更多的支持,引领行业发展。主要包括以下几点:①对国内高端制剂研发企业提供政策支持:解决研发过程中的卡脖子问题,如分段生产的监管问题,以促进国产高端制剂仿制药对原研药的替代。②建议构建高端制剂研发基地或合作平台,与学术界、高端制剂行业全面联动,通过设立研究课题、研习班、线上线下研讨会、培训班等多种形式,监管机构和学术界、行业的充分互动,提升行业对高端制剂的理解,进而加快高端制剂研发。利用药品监管机构在药品开发领域的专业性以及号召力,联合学术界、产业界合力打破高端制剂开发过程中面临的挑战,推动监管机构从监管者身份向高端制剂行业引领者身份的转变,利用其丰富的审评经验,引领高端制剂的开发。③推动高端制剂的一致性评价,全面提升高端制剂仿制药质量。因为高端制剂中的吸入剂、透皮贴剂尚未启动一致性评价,已上市的吸入剂、透皮贴剂目前没有申报一致性评价的路径;而按照新的化学药品注册分类获批的吸入剂、透皮贴剂等仿制药又视同通过一致性评价[20]。这样就导致通过仿制药路径批准的产品视同通过一致性评价,已上市的产品因一致性评价路径不开放而无法通过一致性评价,最终导致市场准入层面产生不公平。④对高端制剂所用的功能性辅料生产企业以及关键设备研发企业给予相应的支持政策。
4.4 完善高端制剂改良型新药的临床优势要求
绕过原研药的专利、突破制备工艺技术封锁、打破功能性辅料垄断等壁垒,在原研高端制剂的基础上开发改良型高端制剂,是打破原研药垄断、增加用药选择、满足用药可及性和可支付性、提高患者医疗水平、降低患者医疗支出的最有效途径。
尽管开发高端制剂仿制药是解决临床用药可及性的主要路径,但在实际开发中却受到种种制约,主要有以下几方面:①以高端制剂中的复杂注射剂仿制药开发为例,仿制药要与原研药达到辅料种类和用量相同,就得使用相同的辅料,而原研药使用的关键辅料往往是其自制或者专供的,仿制药公司无法获得。②原研药的处方还在专利保护期内,仿制药公司无法使用开发相同处方的仿制药。③原研药为提高仿制药开发难度,在药品监管部门制定仿制药生物等效性指南时,提出过高要求,增加了仿制药开发难度。由于上市种种限制和技术壁垒,开发改良型高端制剂是解决这类药品用药可及性的唯一途径。
《化学药品改良型新药临床试验技术指导原则》要求改良型新药要具有临床优势,“临床优势即患者未被满足的临床需求。在目标适应症中,对比已有的标准治疗,新药或新的治疗手段可显著提高疗效;或在不降低疗效的同时,显著降低当前用药患者的不良反应或用药的相关风险,或显著提高患者用药依从性”。在考虑未被满足的临床需求时,不仅要考虑临床优势,还需要考虑临床用药的可及性,应将增加临床用药选择、提高可及性也作为支持其获批的关键。针对临床可及性差的高端制剂,建议将“增加用药选择”和“满足临床用药可及性”纳入高端制剂改良型新药的临床优势范畴,允许通过等效或非劣效临床试验支持其获批上市。
5 总结
高端制剂具有传统制剂不具备的临床优势,能够满足传统制剂无法满足的临床需求,推动其研发上市及市场准入对优化我国医药产业结构以及满足我国公共卫生的需求有着重要的意义。本文通过梳理国内外高端制剂开发的现状,总结现阶段存在的问题,并提出建设性的建议,为推动高端制剂上市许可及市场准入提供参考。相信在多重鼓励、支持、完善的配套措施下,我国高端制剂的研发水平和质量会不断提升,推动高端制剂达到国际先进质量标准,促进我国制药工业长足发展,进一步提升高端制剂的可及性,使人民群众有药可用,更有好药可用!
