男性幼儿双下肢无力1周伴眼睑下垂3 d
2022-08-23郎长会田茂强束晓梅
郎长会 田茂强 束晓梅
(贵州省儿童医院/遵义医科大学附属医院小儿内科,贵州遵义 563003)
1 病例介绍
(1)病史:1岁7个月男童,因双下肢无力1周,伴眼睑下垂3 d入院。1周前因不能行走、不能站立,逐渐加重到不能独坐。入院前3 d出现眼睑下垂,无晨轻暮重,无持物不稳,伴阵发性哭闹、烦躁不安、精神差及思睡。病程中无饮水呛咳及声音低微,无声嘶及吞咽困难,无发热、气促及呼吸困难,无呕吐、抽搐及大小便障碍。门诊以“重症肌无力(全身型)可能性大”收入院,入院当天完善相关检查并对症治疗,第2天因考虑为免疫性疾病开始予静脉注射免疫球蛋白(intravenous immunoglobulin,IVIG)治疗,入院第3天(病程第10天)出现阵发性面色青紫、呼吸暂停及呼吸节律不规则,血红蛋白氧饱和度(oxygen saturation of hemoglobin,SO2)下降至80%,考虑合并中枢性呼吸衰竭转入儿童重症监护室(pediatric intensive care unit,PICU)治疗。
(2)既往史、个人史、家族史:起病前1月余接种水痘及脑膜炎相关疫苗,前1周有呼吸道感染病史。既往生长发育里程碑正常,入院时可讲1~2个字,认知能力正常。患儿系第1胎第1产,出生史及家族史无异常。
(3)入院时体格检查:T 36.8℃,P 130次/min,R 33次/min,SO298%,嗜睡状,精神差,哭闹不安,不能独坐,双侧眼睑平视时遮挡角膜位于9~3点方向,眼球运动体格检查不配合,双侧瞳孔直径3 mm,光反射灵敏,咽反射存在,颈软,心肺腹体检未见异常,双上肢肌力4级,双下肢肌力3级,肌张力正常,双侧腱反射消失,布氏征、克氏征阴性,共济运动体格检查不配合。
入院第3天转入PICU时体格检查:T 36.5℃,P 150次/min,R 20次/min,SO294%(气囊加压给氧下),镇静状,呼吸节律不规则,时有呼吸暂停,双侧瞳孔正圆等大,直径约5 mm,瞳孔对光反射消失。双上肢肌力约3级,双下肢肌力约2级,双侧腱反射消失,病理征阴性。
(4)实验室检查:血常规、电解质+肝肾功能、血糖、C反应蛋白、肌酶、血氨、乳酸、同型半胱氨酸正常。入院当天(病程第7天)肌电图未见异常,神经传导速度(nerve conduction velocity,NCV)无明显异常。入院第2天(病程第8天)脑脊液常规、生化未见异常。2次新斯的明试验均阴性。头颅CT、MRI未见异常。
2 诊断思维
病例特点:(1)幼儿期起病,急性进展性病程,起病前有疫苗接种及呼吸道感染病史;(2)肢体无力进行性加重、眼睑下垂;(3)病程第5天出现脑病表现(嗜睡);(4)病程第10天出现中枢性呼吸衰竭症状(呼吸暂停、呼吸不规则、发绀、SO2下降至80%);(5)既往运动、语言发育正常。入院时体格检查:嗜睡状,双上肢肌力4级,双下肢肌力3级,双侧腱反射消失。辅助检查:肌电图正常,NCV未见明显异常。
首先定位。(1)下运动神经元,包括前角细胞、周围神经、神经肌肉接头、肌肉等。①前角细胞:患儿呈驰缓性肢体无力表现,需考虑前角细胞损害,如肠道病毒感染等,但患儿瘫痪呈对称性,肌电图未提示神经源性损害,暂时不支持前角细胞病变,但需进一步行脊髓MRI协助排除。②神经肌肉接头:患儿以双下肢无力、眼睑下垂起病,需考虑有无全身型重症肌无力,但症状无波动性,无晨轻暮重现象,2次新斯的明试验均阴性,不支持。③肌肉:肌酶正常,肌电图未提示肌源性损害,可排除。④周围神经:患儿以双下肢无力、眼睑下垂起病,四肢肌力下降,腱反射消失,高度怀疑周围神经性病变,但患儿NCV未提示潜伏期延长、NCV减慢或动作电位波幅下降及F波异常,也可能在病情早期尚未出现异常,故需进一步动态观察病情,适时复查NCV。(2)上运动神经元,包括脊髓、大脑等。①脊髓锥体束:因患儿有肢体无力、腱反射消失等表现,需考虑脊髓休克期,但患儿无大小便障碍,且患儿年龄偏小,无法配合感觉体格检查,进一步查颈胸腰段脊髓MRI协助诊断。②脑干:因患儿除肌无力外还伴有无法解释的嗜睡,当时无明显呼吸困难、呼吸无力及缺血缺氧等表现,数天后才出现中枢性呼吸衰竭(呼吸暂停、呼吸不规则及瞳孔散大),需考虑是否累及脑干等。
其次定性。神经系统疾病定性包括感染性炎症、免疫性炎症、遗传性、代谢性、肿瘤性、血管性、中毒性及外伤性等。结合本例患儿急性起病,呈进行性加重,考虑感染性或免疫性炎症,需排除遗传性及血管性、中毒性、外伤性病因等。但患儿本次病程中无发热,辅助检查炎症指标不高,故不支持感染性炎症。急性起病,既往运动、语言发育正常,辅助检查无明显代谢紊乱,遗传代谢性疾病可能性小。患儿急性起病,有眼睑下垂及肢体无力,但患儿头颅及脊髓MRI均未见异常,不支持肿瘤性病因。血管性、中毒性、外伤性病因均缺乏相应病史,暂不考虑。本例患儿病前有疫苗接种及前驱感染史,需高度怀疑免疫性炎症所致,需行相关血及脑脊液抗神经节苷脂抗体(anti-gangliosides antibody,AGA)检查[1],并进一步动态监测NCV及脑脊液相关指标变化。
3 进一步检查
入院第3天(病程第9天)复查NCV提示双侧胫神经H反射消失,运动、感觉NCV及F波正常。入院1周(病程第13天)复查脑脊液,提示细胞数仍正常,蛋白增高至1 282 mg/L(参考值200~400 mg/L),提示蛋白细胞分离。同时,外送血清及脑脊液相关抗体,包括水通道蛋白4、少突胶质细胞糖蛋白、星形胶质纤维酸性蛋白、AGA检测(抗GQ1b抗体IgG和IgM、抗GD1b抗体IgG和IgM,以及抗GM1抗体IgG和IgM),结果显示血清抗GD1b抗体IgG阳性,脑脊液抗GD1b抗体IgG阴性,余均为阴性。颈胸腰段脊髓MRI未见异常。
4 诊断及诊断依据
诊断:吉兰-巴雷综合征(Guillain-Barré syndrome,GBS)、Miller-Fisher综 合 征(Miller-Fisher syndrome,MFS)及Bickerstaff脑干脑炎(Bickerstaff brainstem encephalitis,BBE)重叠综合征。诊断依据:(1)GBS:患儿有前驱感染,双下肢无力进行性加重,在2周达高峰;腱反射消失;脑脊液蛋白细胞分离。(2)不完全性MFS:突出表现的眼外肌麻痹,无其他颅神经受累,伴腱反射消失,无明显共济失调表现;伴有脑脊液蛋白细胞分离现象,血清抗GD1b抗体IgG阳性,诊断为不完全性MFS。(3)BBE:眼外肌麻痹;原发病不能解释的脑病样表现(嗜睡);中枢性呼吸衰竭(呼吸节律异常)。
5 临床经过
经IVIG[400 mg/(kg·d)×5 d]、甲泼尼龙冲击[20 mg/(kg·d)×3 d]、5次血浆置换、呼吸机辅助通气及对症支持治疗,患儿病情逐渐好转(眼睑下垂及嗜睡好转),精神可。出院时体格检查:神志清楚,呼吸规则,双上肢肌力3级,双下肢近端肌力2级,远端肌力3级。出院后康复治疗半年,患儿可独立行走、无眼睑下垂及眼球活动障碍。末次随访时(出院后1年),患儿运动基本恢复,行走有力,可跑跳,可上楼。体格检查示眼球活动正常,无眼睑下垂,四肢肌力及肌张力正常,腱反射仍未引出。复查NCV示H反射恢复正常。
6 讨论
GBS、MFS及BBE重叠综合征可同时出现周围神经及脑干损害,临床异质性强。GBS是由感染后引起的免疫性多发性神经病变,其临床特征为急性起病、快速进行性对称肌无力和腱反射减退或消失[2]。眼外肌麻痹、共济失调和胰反射消失认为是MFS的典型三联征[3]。GBS和MFS均为感染后急性单相性免疫性疾病,且易伴脑脊液蛋白细胞分离,但二者又有不同的核心临床症状。单纯性MFS,单相性病程,4周内达疾病高峰,有典型三联征,无肢体无力或中枢神经系统受累,并除外其他疾病,脑脊液蛋白细胞分离现象为支持条件。然而,不是每个患儿都有典型三联征[4],如果缺乏典型三联征,伴血清AGA阳性及脑脊液蛋白细胞分离,提示不完全性MFS,表现为:(1)不伴共济失调称为急性眼外肌麻痹;(2)不伴眼外肌麻痹称为急性共济失调神经病;(3)仅表现眼睑下垂称为急性眼睑下垂;(4)仅表现瞳孔散大称为急性瞳孔散大[5]。BBE以共济失调、眼外肌麻痹、腱反射消失及脑病为特征,可累及脑干和周围神经系统。BBE与MFS有一些共同特征如共济失调、眼外肌麻痹等。BBE除了上述症状外,伴有脑病样症状如嗜睡或意识障碍、出现椎体束征[6-7]。本例患儿有典型GBS的临床表现,如下肢无力进行性加重逐渐累及上肢、腱反射消失、脑脊液蛋白细胞分离,但临床上还有不能用GBS解释的症状及体征,如眼外肌麻痹,但缺乏后组颅神经麻痹的表现(无声音变弱、饮水呛咳及吞咽困难等),考虑重叠MFS;因患儿无明显的共济失调表现,考虑为不完全性MFS。此外,患儿有明显的脑病样表现(嗜睡)且不能用其他原因解释(当时无明显呼吸困难、呼吸无力及缺血缺氧等表现),考虑重叠BBE。
文献报道外周血AGA在儿童GBS谱系疾病中多以血清抗GQ1b IgG抗体为主,可表现为GBS、MFS及BBE重叠[8-9],较少有抗GD1b抗体阳性的报道。进一步在PubMed(https://pubmed.ncbi.nlm.nih.gov/)或Gene Medical(https://www.geenmedical.com/)中以“GD1b IgG”“GD1b IgM”“children”为关键词,检索到5篇文献,共5例患儿[10-14],以男性为主(4/5),多在学龄期起病(年龄5岁5个月至12岁)。临床表现有肢体无力(2/5),少数出现言语困难、构音障碍及眼外肌麻痹。腱反射正常或减退。辅助检查可出现脑脊液蛋白细胞分离,神经电生理检查可出现NCV受限、轴索变性或正常;AGA检测均有抗GD1b抗体阳性。所有患儿均接受糖皮质激素或IVIG治疗(表1)。由此可见,在抗GD1b抗体相关疾病的临床表现和神经电生理表现存在极大异质性。而本例患儿仅抗GD1b IgG抗体阳性,且临床表现较文献中患儿表型更重。也有文献报道单抗体阳性患者较多抗体阳性患者的临床表现更重[15]。Kaida等[15]分析同时存在2种或2种以上AGA在细胞膜上形成神经节苷脂复合物,这种神经节苷脂复合物对GD1b抗原的结合活性弱于只有1种抗GD1b抗体的结合活性。这可能是该患儿症状比其他患儿更严重的主要原因。因此,只有1种抗GD1b抗体在预测疾病严重程度方面具有重要意义。
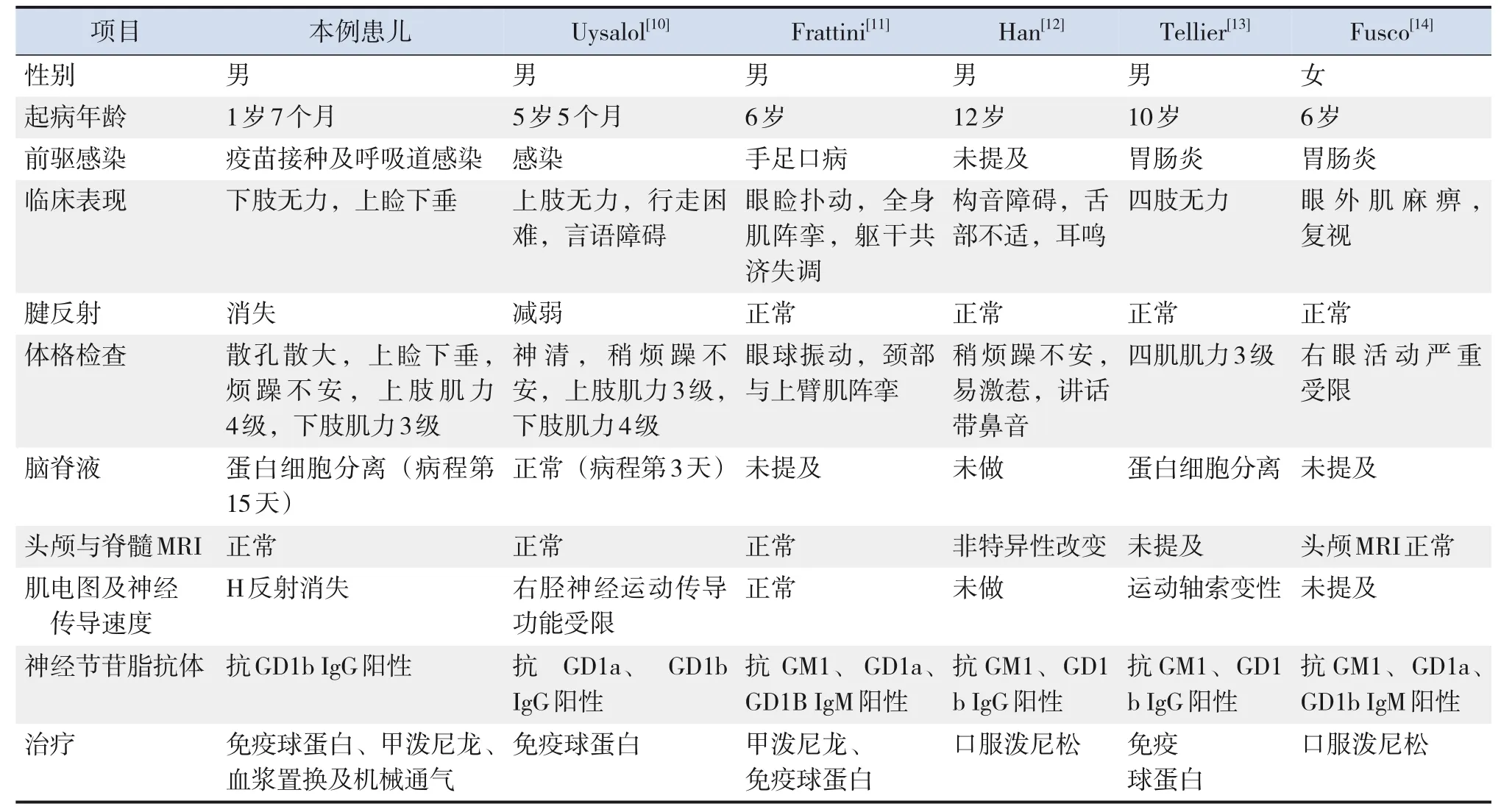
表1 儿童抗GD1b抗体阳性患儿的临床表型
Kaida等[15]报道成人抗GD1b抗体阳性患者多数以共济失调和上睑下垂起病。本例患儿有上睑下垂,但无明显共济失调,分析该患儿的共济失调可能被嗜睡等症状所掩盖。
本例患儿H反射消失,可能与AGA引起脊髓背根神经元中肌梭或Ⅰa类神经元受损有关,并未引起脱髓鞘或轴突病变,这就可解释患儿运动/感觉NCV正常,潜伏期或复合肌肉动作电位正常,提示其更容易恢复。Kuwabara等[16]报道约70%的MFS患儿中观察到H反射缺失。MFS患儿中腓肠神经活检显示郎飞结延长,未提示脱髓鞘,可能出现NCV和F波正常,与本例患儿一致。朱莹等[17]观察典型GBS及MFS电生理区别,发现典型GBS约有一半表现为运动伴感觉神经传导异常或单纯运动神经传导异常(27.8%),而MFS组约1/3表现为单纯H反射异常,1/3表现为单纯感觉传导异常,还有1/3为正常肌电图。Rasera等[18]和Dachy等[19]均报道在所有MFS患儿中均有双侧H反射缺失。以上研究结果提示双侧H反射消失在MFS患儿中是一个较灵敏的指标。
GBS、MFS及BBE重叠综合征的治疗包括IVIG或糖皮质激素、血浆置换、康复及对症治疗[4]。关于糖皮质激素治疗目前无统一标准,对重症病例,建议大剂量静脉滴注甲泼尼龙[20~30 mg/(kg·d)]3~5 d,然后改为口服泼尼松[1 mg/(kg·d)]维持4~6周[20]。本例患儿经IVIG、血浆置换、糖皮质激素及康复等治疗后,半年内完全恢复基线水平。
目前认为GBS、MFS及BBE重叠综合征总体预后较好,约80%的此类患儿在发病6个月后恢复独立行走能力,3%~10%的患儿死亡,死亡原因最常见是呼吸系统及心血管并发症[2]。
7 结语
目前临床医生对于儿童GBS、MFS及BBE重叠综合征(尤其在症状不典型时)认识不足,且该病有潜在生命危险,若延误诊断可导致患儿出现呼吸衰竭等严重并发症。当患儿出现四肢无力、上睑下垂及嗜睡时,应怀疑GBS、MFS及BBE重叠综合征。应尽快进行NCV检查,重点关注有无H反射,并完善AGA,早期诊断及治疗可改善预后。
利益冲突声明:所有作者均声明不存在利益冲突。
