38例临床患者留置导尿管的微生物群落多样性分析
2022-06-28陈豪特杨小琴
李 丁,陈豪特,杨小琴,王 忠,甘 辛
(1.中国中医科学院西苑医院 泌尿外科,北京 100091;2.国家食品安全风险评估中心,卫生部食品安全风险评估重点实验室,北京 100021)
研究显示所有到院就医的感染类疾病中,泌尿道感染占比较高,感染率高达20%~30%,并于全年龄段和性别可见,在为患者带来严重的躯体症状的同时也极大的增加了疾病治疗的经济成本[1-2]。除社区感染外,泌尿道感染作为最常见的院内感染之一也是仅次于呼吸道感染的第二大院内感染,受到世界各国广泛关注[3]。美国每年仅儿童泌尿道感染的治疗就需要医保系统投入1.8亿美元的费用,同时每年还需要约150万名临床医生参与诊治[4]。留置导尿管是临床解决排尿困难的有效技术手段,值得注意的是泌尿道感染与医疗机构使用导尿管密切相关,原因包括无菌操作不当、插管过程中损伤尿道粘膜、留置时尿道口未消毒或消毒不严格等。留置导尿管为细菌侵入人体增设了通道,一旦其表面出现细菌定植并生成生物膜将进一步增加患者感染的风险,报道显示我国因导尿管引发的患者泌尿道感染率可达6.68%~23.5%[5-6]。
本研究以16S rRNA测序为手段,该方法通过PCR扩增样品中16S rRNA片段后对其中的细菌进行目、属、种水平的鉴定,具有读长高、精度高、通量高以及无偏性等优势,作为一种成熟可靠的方法被广泛应用于菌种鉴定和微生物群落多样性的研究[7]。相较于微生物传统的分离培养方法,该方法不受培养方法的制约,可以更好的发现样本中的罕见细菌、难以培养以及不具有典型特征形态和生化反应的细菌。通过检测38例北京市西苑医院2020年11~12月份间收治的临床患者短期和长期留置的导尿管微生物群落构成,旨在研究导尿管留置可能给患者造成的感染风险,并为下一步指导临床干预提供可靠的数据支持。
1 材料与方法
1.1 样本来源 本研究以北京市中国中医科学院西苑医院泌尿外科2020年11~12月收治的38例导尿管留置患者为对象开展,并对患者临床资料进行回顾性分析。38例患者中,男性31例,女性7例,年龄64~99岁,平均年龄为(82.1±7.2)岁,导尿管平均留置时间为(25.50±9.66)d。所有入组病例均排除2周内抗生素、激素及免疫抑制剂等可能对细菌感染存在较大影响的药物应用史。37例患有因前列腺增生或神经源性膀胱等造成的尿潴留,占全部病例的97.4%。其中20例留管过程中有过抗生素、中药等在内的药物干预,占52.6%,其中八宝丹、癃清片、临床中药饮片、头孢西丁、头孢他啶、哌拉西林钠他唑巴坦为主要干预用药。样品留置时间7~41 d,以14、21、28、35 d为分组节点(见表1),每组样品数分别为S1(6),S2(5),S3(7),S4(15),S5(5)。导尿管为一次性使用无菌导尿包-“抗菌超滑行型”(山东百多安医疗器械有限公司)注册证号:鲁食药监械(准)字2014第2660506号。
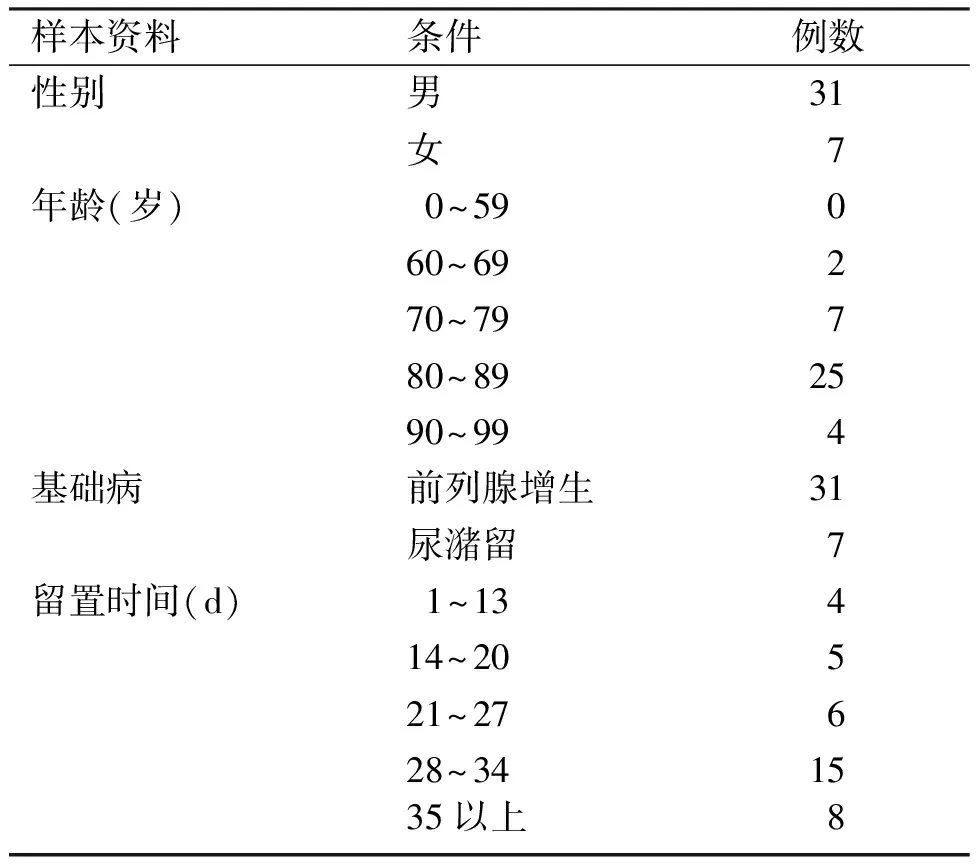
表1 38份样本信息统计
1.2 样本采集 取样前先对患者尿道口进行消毒,戴无菌手套取出导尿管,用经灭菌处理的剪刀截取导尿管留置于患者体内部分即尿管自远端至水囊部分,放置于(5×5)cm无菌规格板上。使用灭菌医用棉签拭子于尿管尖端外周表面不同部位紧贴尿管表面擦拭不少于5次,将拭子尖端折断并置于含有保存液的灭菌Corning管中保存。
1.3 文库构建和测序 DNA提取采用Qiagen微生物DNA纯化试剂盒(NO:50214)。16s rRNA测序采用V4+V5特异性引物515FB:5′-GTGYCAGCMGCCGCGGTAA-3′,926R:5′-CCGYCAATTYMTTTRAGTTT-3′。PCR条件为预变性95℃,180 s,变性98℃,20 s,退火55℃,15 s,延伸72℃,15 s,循环25次,终延伸72℃,60 s,反应体系为KAPA 热启动高保真酶(NO:KK2602)50 μL体系。获得的PCR产物进行1%琼脂糖凝胶电泳检测DNA无降解后切胶回收,胶回收条件依照Omega D2500-02胶回收试剂盒。文库经AglientBioanalyzer和Qubit检测定量合格后上机测序。测序平台选择Hiseq 2500,由北京博奥汇玖生物科技有限公司采用PE250模式进行,对样本16s rRNA基因 V4-V5区进行测序分析。
1.4 数据分析 序列下机后经skewer软件进行数据过滤,并使用FLASH软件对paired-end reads进行拼接[8-9]。拼接后的序列经CD-HIT生成OTU并进行OTUs的聚类分析,在OTUs聚类的基础上使用SILVA数据库对每个OTU的代表序列进行物种注释,从而获得样本的物种注释信息及基于物种的丰度分布情况[10-11]。对OTUs同时进行Alpha多样性、丰度等在内的相关分析,获取样品内物种丰富度、均匀度信息和不同样品间OUTs的共性和特性信息等。此外,对OTUs进行多序列比较构建系统进化树,基于进化树信息和丰度信息进行Beta多样性分析,获得不同样品和分组的群落构成差异的信息,以PCA、NMDS和PCoA及距离矩阵热图等方式进行展示。
2 结果
2.1 DNA提取和文库构建及序列检测 样品经提取后DNA总含量经Qubit双链试剂盒定量在57~3 850 ng,说明不同样品的微生物总量有较大差异。1%琼脂糖电泳进行快速检测,结果显示DNA 无降解。扩增后产物使用Qubit进行建库前DNA 浓度测定,前一步得到的DNA 浓度的Qubit测定值最低为3.34 ng/μL,使用55.5 μL 体系建库计算,有0.185 μg 总量,满足最低0.1 μg 的建库需求。文库构建后经AglientBioanalyzer2100进行检测,其中样品的主峰长度574 bp/573 bp/576 bp,除去126 bp 的接头序列和24 bp 的分子标记序列后,剩余期望长度为424 bp/423 bp/426 bp,与16S rRNA基因V4~V5区的期望长度412 bp 接近,考虑到GC 含量等因素的影响以及毛细管电泳本身的误差,可以认为所扩增片段的长度与期望值基本相符。
2.2序列检测 样品混合后定量至12pm进行上机测序。根据样品提取有效测序,序列中含有特异性扩增引物序列,长度大于可供分析标准的序列称为有效序列。共获得1 609 398条序列,其中有效序列1 603 007条,说明整体测序质量优良。单个样品平均总序列42 353条,平均有效序列42 184条,平均有效率达到99.61%。最少样品有效序列有35 390条,最多样品有44 991条,说明样品均一度较好。
本研究样品的目标区域16S rRNA基因V4~V5区的平均期望长度为412 bp。双端测序正反向读取的序列总长为500 bp,扣除分子标记24 bp,共有476 bp。有效序列长度在382 bp至442 bp之间。测序所得序列中占比最多的5种长度及其百分比分别为:412(50.44%),413(24.46%),411(8.35%),414(6.13%),408(5.59%),其余长度占比为5.03%,与期望占比长度相符。
2.3 OTU序列聚类分析 根据序列相似性,按97%的相似度,与Silva v132数据库进行比对,将序列归为多个 OTU(操作分类单元)进行分析,共得到11 350个OTUs和1,668,076个优质序列。
2.4 Alpha多样性分析 当序列达到2 000左右时香浓曲线进入拐点达到平台期(见图1),表明本次测序已经覆盖样品物种序列,测序量饱和能够展现样本细菌的多样性和分布,即使增加深度也不会对样品多样性产生影响。相较于肠道细菌、皮肤细菌、土壤细菌等自然测序群体,本实验样品多样性达到饱和所需序列较少,说明导尿管为相对封闭环境,微生物群落具有特定性。Rank Abundance曲线在水平方向上有较大跨度,垂直方向也具有分离,说明样本具有多样性。
图1 38份样品香浓曲线
2.5 物种注释分析 利用SILVA数据库对16s rRNA基因测序数据分别在界、门、纲、目、科、属、种进行注释根据样本中包含的各水平物种间的距离进行水平物种聚类分析(见图2)。16s rRNA测序结果显示,以S1-S5时间段分组,各组样品中革兰氏阴性菌所占比例均高于阳性菌。在门水平上,各样品的优势菌分布随时间变化具有规律性。以28 d为分界点,梭杆菌门(Fusobacteria)丰度上升,28 d以上样本以35 d为分界可发现厚壁菌门(Firmicutes)和变形菌门(Proteobacteria)丰度发生规律性变化。28 d内以14 d为分界点可发现梭杆菌门随时间增长比例上升,其中14~21 d组合21~28 d组在热脱球菌(Deinococcus-Thermus)丰度上具有数值差异。样本中α-变形菌纲(Alphaproteobacteria)丰度短期(7~14 d)高于中长期留置患者(见图3)。其中36个样品存在含量>50%的优势菌门,占全部样品的86.8%,其中19个样品优势菌门为变形菌门,含量63%~100%,占52.8%(19/36),12个样品优势菌门为厚壁菌门,含量54%~100%,占33.3%(12/33),3个样品优势菌门为放线菌门,含量67%~81%,占8.3%(3/36),2个样品优势菌门为梭杆菌门,含量53%~78%,占5.6%(2/36)。S1-S5各时间段分组显示,S1中变形菌门占62.3%,厚壁菌门占33.8%;S2中变形菌门占46.7%,厚壁菌门占36.3%;S3中变形菌门占46.6%,厚壁菌门占34.2%;S4中变形菌门占42.0%,厚壁菌门占34.0%;S5中变形菌门占73.5%,厚壁菌门占16.0%(见图4)。样品测序结果中包含5种常见的院内感染细菌,其中肠球菌是主要检出菌,阳性率为68.4%,大肠埃希菌阳性率为47.4%,铜绿假单胞菌阳性率为39.5%,金黄色葡萄球菌阳性率为21.1%,肺炎克雷伯菌阳性率为2.6%。5个样品肠球菌占比>53%,其中3个样品肠球菌占比>96.0%,留管时间除1例为7 d外其余均≥28 d,5例患者中3例患者服用癃清片2例未使用药物干预。另有3个样品铜绿假单胞菌占比>73.7%,留管时间≥21 d,其中2例服用头孢西丁或哌拉西林1例患者未使用药物干预。
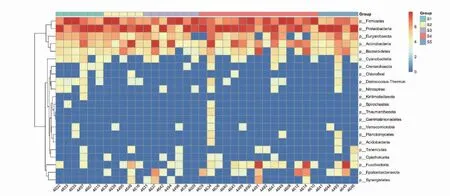
图2 导尿管留置时间与微生物群落聚类分析

图3 38份样品中α-变形菌纲分布信息

图4 38份样品微生物群落结构
2.6 分组分析 Anosim,PCA,PCoA,NMDS,MRPP和Turkey分析均为检测到组间显著差异。但是针对各组和菌群的LEfSe检测发现,组1与孪生球菌属(Gemella),拟甲色球藻属(Chroococcidiopsis),寡养单胞菌属(Stenotrophomonas)有较高关联(LDA Score log10>2),组2与乳球菌属(Lactococcus),组5与微单胞菌属(Parvimonas)有较高关联。
3 讨论
导尿管是引发下尿路感染临床病例的重要因素之一,病情加重时还可以诱发上尿路感染继而发生慢性肾功能衰竭、继发性高血压等严重并发症,近年来如何控制导尿管引发的泌尿系感染已成为临床关注的重点。目前尚未见国内外应用16s rRNA高通量测序方法展示患者导尿管表面病原微生物菌群多样性及分布特点的其他相关报道。该方法由于不受培养方法的制约,可以更加深入全面的发掘患者潜在的感染风险,为今后指导临床抗感染治疗提供了详实可靠的数据。
留置尿管是一个相对较为封闭的环境,这在宏基因组研究中是较为特殊的。根据本研究Alpha多样性结果分析显示,当序列条数达到2 000左右时上升曲线便出现拐点并进入平台期,表明检测在较小的通量时就达到饱和,这相对于肠道、土壤等半开放环境都是差异较大的。本研究探索的检测通量也有助于未来相似研究确定通量范围、降低检测成本、开发检测工具。超高的通量也保证了本研究测序深度和广度较为全面的覆盖了样本的所有物种,检测也比较均匀。
本研究检测的38例样品中包含了包括肠球菌、大肠埃希菌、铜绿假单胞菌、金黄色葡萄球菌和肺炎克雷伯菌在内的临床上常见的5种院内感染细菌,其中肠球菌、大肠埃希菌、铜绿假单胞菌和金黄色葡萄球菌污染率≥21.1%,表明留置尿管微环境对于感染风险有影响。这与陆金金等报道的2012—2015年尿路感染病原菌主要为大肠埃希菌(37.1%),粪肠球菌(19.9%),铜绿假单胞菌(8.0%),肺炎克雷伯菌(7.5%),白色念珠菌(5.2%)及颜复生等2009年报道的647株尿路感染分离株主要为大肠埃希菌(40.8%)、念珠菌属(28.2%)、肠球菌属(7.9%)、链球菌属(4.9%)、克雷伯菌属(3.6%)结果相似[12-13]。38份样品中68.4%存在肠球菌,其中3份长期留置的样品中该菌占比>96%。肠球菌广泛分布于自然环境中,可寄居于人和动物的肠道内,是引起院内感染的重要条件致病菌,能引起尿路感染、脑膜炎、心内膜炎和脓毒血症等在内的多种感染性疾病。其发病率近年来呈现上升趋势。研究表明,院感肠球菌具有耐药率高,存在多重耐药等特点,可能成为耐药基因的储存库,因其固有抗生素耐药性及其迅速获得额外抗生素耐药性的能力使感染很难治疗,特别是万古霉素耐药菌的出现,构成了重大的感染控制负担[14-15]。铜绿假单胞菌也是临床最常见病原菌之一,本研究中39.5%的样品存在该菌,其中3份长期留管样品中含量>73.7%,该菌本身携带染色体介导的AmpC β-内酰胺酶,可对氨苄西林、阿莫西林、氨苄西林-舒巴坦、阿莫西林-克拉维酸、头孢噻肟和头孢曲松产生天然耐药,同时其外排泵机制可将β-内酰胺类、氯霉素、复方新诺明、四环素类及替加环素等泵出胞外,从而产生耐药,该菌多重耐药发生率较高[16]。目前大量研究显示,肠球菌、铜绿假单胞菌、大肠埃希菌、金黄色葡萄球菌和肺炎克雷伯菌均可以携带可传递的耐药质粒,一旦发生质粒传递导致的多重耐药将进一步加重病人的负担和临床治疗的难度,也对其他住院病人的健康产生极大威胁[17]。8例单一细菌占比超过53%的病例中,5人口服中药或抗生素药物干预,3例未服用任何药物未见差别(P>0.05),结果显示口服药物干预与导尿管菌落构成高度单一没有相关性,可能与药物在取样位置难以达到抑菌所需浓度有关。目前研究也指出生物膜与尿路感染具有密切关系,细菌通过多糖蛋白复合物将细菌包裹其中附着在病灶或导管表面[18]。本研究检出的大肠埃希菌、铜绿假单胞菌、金黄色葡萄球菌、肺炎克雷伯菌等细菌均具有形成生物膜的能力,长期留置导尿管的患者因为尿道缺少尿液盥洗作用,导致细菌容易黏附在导尿管和尿路上皮形成生物膜,而生物膜可以增强细菌耐药性和逃避宿主免疫,常规消毒剂也难以彻底清除生物膜,从而引发持续性的感染。
留置尿管封闭环境的16s rRNA检测显示出与开放环境不一样的规律。从个体层面来看个体间差异较大,极端个体较多,在分组层面上,极端个体的出现使得统计上分组特征规律有限。在门水平上,高于85%的样品出现主导门超过50%的情况,多个样品在属甚至种水平上优势菌超过90%,说明这一现象不是偶然的。这表明在无菌封闭环境,优先定殖菌可获得巨大的生态优势,从而导致极端占比的频繁出现。优势定殖菌具有一定的规律范围,但就具体菌种的定殖有一定的随机性,可能与病人的身体状况、饮食、用药具有关联。通过对全部样品的聚类研究发现,样品菌群变化规律总体上与留管时间相关。长留置时间与高度单一菌群结构有伴随性出现,同时某些短期留置含量较低的细菌随着时间逐渐升高,说明长时间单一环境对部分菌群具有筛选并易导致感染风险。本研究结果显示,需要进一步增加对尿管留置病人的病例研究,特别是对需较长时间留置尿管病患尿管更换周期的研究应纳上日程,同时通过大量样本收集和数据分析对留置尿管中的优势菌种进行识别和检测,为尿管留置导致感染的早期治疗和针对性治疗提供详实可靠的数据支持。
