纤毛通路基因罕见危害性基因变异对人腰骶部神经管畸形的致病性
2022-05-12蔡春泉陈晓丽
汪 静,刘 芳,谢 华,蔡春泉,姜 宏,陈晓丽
神经管畸形(neural tube defect,NTDs)是一类由于胚胎发育早期神经管闭合失败或闭合不完全所致的严重出生缺陷。据统计,NTDs的全球发病率为0.5‰~2%,中国的发病率接近2.74‰[1]。NTDs是一类由环境和遗传因素共同作用导致的复杂性疾病。目前通过人群及小鼠敲除模型研究已发现400多种可能导致神经管畸形的基因[2],其中热点基因包括CELSR1、FZD6、PRICKLE1、DVL2、VANGL1、VANGL2等极性基因[3]。近年研究[4]认为新生突变是导致出生缺陷及神经发育疾病的常见原因。2015年,Lemay et al[5]对43个散发NTDs家系研究也证实新生突变是导致人腰骶部NTDs发生的原因之一。
纤毛是突出于细胞表面的微小细胞器并广泛存在,与人类发育健康和疾病如麦克尔综合征(Meckel syndrome,MKS)、Joubert综合征(Joubert syndrome,JS)等密切相关。上述综合征包括胎儿脊柱裂、脑脊膜膨出、脑组织缺如等与NTDs表型类似的临床表现。但关于纤毛基因变异与人NTDs的相关性研究较少,本课题组前期研究[6]证实:同时参与纤毛发育的极性基因PARD3突变与人NTDs相关,且均为颅部NTDs。鲜有关于纤毛基因与人类腰骶部NTDs的相关性研究。该研究利用AmpliSeq技术设计了包括极性基因(如PARD3)及明确纤毛病候选基因在内的49个纤毛相关基因的编码区靶向捕获Panel,对56个散发腰骶部NTDs家系进行测序,以期明确纤毛罕见危害性变异对人腰骶部NTDs发生的致病性。
1 材料与方法
1.1 研究对象选择在首都儿科研究所和天津市儿童医院明确诊断的腰骶部神经管畸形患者56例,均无家族史,其中男性36例,女性20例。所有腰骶部神经管畸形患者由临床医师评估并影像学辅助诊断,最终手术明确诊断。全部患者父母亦由临床医师评估结合影像学辅助诊断无神经管畸形。本研究通过首都儿科研究所伦理委员会审查,所有患儿均由其父母或监护人签署知情同意书。
1.2 主要仪器及试剂Qubit 2.0 荧光光度计、Ion OneTouch 油包水PCR仪、Ion OneTouch ES 仪、Ion Torrent PGM测序仪(美国Life Technologies 公司)。全血DNA提取试剂盒(美国Qiagen公司);Agencourt AMPure XP磁珠化试剂(美国Beckman Coulter公司);Qubit dsDNA HS Assay 试剂盒、Ion PGMTM200 Sequencing 测序试剂盒、Ion 316TM芯片、 Ion Xpress Barcode Adapters1-16试剂盒、Ion AmpliSeq Library 试剂盒(美国Life Technologies 公司)。
1.3 实验方法
1.3.1DNA制备 采集56例神经管畸形患儿及其表型正常父母外周血提取基因组DNA,采用DNeasy Blood & Tissue kit试剂盒(美国Qiagen公司)提取DNA。取2 μl已提取的基因组DNA,应用紫外分光光度仪测量DNA浓度,A260/A280=1.8~2.0,A260/A230>2.0,计算基因组DNA含量。
1.3.2纤毛基因Panel的靶向深度测序 利用既往NTD小鼠模型中发现的纤毛基因[2]、OMIM系统内明确纤毛发育相关遗传病(https://www.omim.org/)及课题组最近发表的文章[6]构建纤毛候选基因列表。采用Ion AmpliSeq Library在线设计系统形成纤毛基因的AmpliSeq靶向捕获和多重PCR扩增试剂盒,之后和Ion Xpress Barcode Adapters试剂进行文库构建,制备的文库片段大小为200~300 bp;接着用Ion OneTouchTM200 Templete 试剂盒在Ion OneTouch上对文库进行乳化,在Ion OneTouch ES上对磁珠进行富集;最后采用Ion PGM 200 试剂盒和Ion 316TM芯片在Ion Torrent PGM平台进行测序。
1.3.3数据分析 实验数据自动传到Ion Torrent服务器,点击已完成的测序,根据要求完成Coverage Analysis和Variant Caller分析,并对测序数据的数据量、平均测序深度、产生的BAM、BAI、VCF.GZ、XLS文件进一步分析。将VCF.GZ文件导入wANNOVAR(http://wannovar.usc.edu/ )软件,分析所获各变异位点在人群的分布频率、氨基酸改变、变异分类(Indel、SNV),优先选择Loss of Function变异,根据错义变异的生物信息学软件(Mutation Taster,PROVEAN,Polyphen-2,REVEL)预测结果确定其危害性。将BAM、BAI文件导入软件IGV,明确各位点测序数据是否为真性突变,排除测序得到的假阳性数据。
2 结果
2.1 测序数据基本情况本研究收集的家系均为1个神经管畸形患儿配伍表型正常的父母,因此首选筛查双位点罕见基因变异、新生基因变异。测序数据与公共数据库[Genome1000、dbSNP、NHLBI、Exome Sequencing Project (ESP)、ExAC Browser]比对,结合神经管畸形的人群患病率,锁定频率小于1%的外显子区非同义单核苷酸突变。对测序数据进行质量控制,测序覆盖率97%~98%,平均测序深度400X~600X。
2.2 NTDs患者存在CRB2复杂杂合变异测序结果显示,1例患儿存在CRB2基因两个突变位点的复杂杂合变异(c.G1392C, p.R464S; c.T3448C, p.C1150R),且均为危害性突变。其中c.G1392C, p.R464S突变位点遗传自患儿母亲,公共数据库中未见报道,另一个突变位点c.T3448C, p.C1150R遗传自患儿父亲,公共数据库中为罕见变异(MAF<1%),见图1、表1,两突变位点在物种间高度保守,见图2。

图1 CRB2复杂杂合突变位点信息A:CRB2基因结构,红色标注分别为患者携带CRB2;b:患者c.1392G>C变异来源于母亲; c:患者c.3448T>C变异来源于父亲;S、M、F分别代表患者、母亲、父亲

表1 突变位点基本信息
2.3 NTDs患者存在GLI3新生突变测序结果显示,1例患儿存在GLI3基因的一个新生突变(c.C580T, p.H194Y),该突变经软件预测为危害性突变,且公共数据库均未见报道,见表1、图3,该突变位点在物种间高度保守,见图2。鉴于该家系发现的GLI3基因变异为新生错义突变,笔者首先通过其他位点的连锁分析证实了患者与父母的亲子关系,用IGV查看家系测序数据,发现GLI3基因片段测序覆盖率及测序深度较低,故采用Primer 3.0针对基因设计引物,对所有家系进行Sanger测序验证,证实了GLI3新生突变位点的真实性,并且在其他55个家系均未见该基因的罕见变异,见图4。其余54个神经管畸形家系尚未发现双位点罕见基因变异及新生基因变异。

图2 突变位点在8个物种间保守性分析
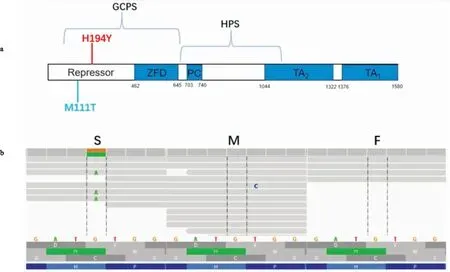
图3 GLI3新生突变位点信息A:GLI3编码蛋白结构,红色标注为本文患者携带GLI3新生突变,蓝色标注为文献[7]中食管闭锁伴有半椎体患者携带GLI3突变;b:panel测序结果显示新生突变;S、M、F分别代表患者、母亲、父亲GLI3是GLI家族锌指结构3。GLI家族锌指蛋白是经典hedgehog(HH)信号通路的转录效应因子,参与胚胎形态发生的各个阶段,并且是组织正常发育的必须调节因子。GLI3基因突变与格雷格头、多指(趾)、并指(趾)综合征(Greig cephalon polysyndactyly syndrome,GCPS)等相关。GLI3突变小鼠因大脑、脊柱、四肢、肺等多器官缺陷无法存活。2014年Yang et al[7]报道1例食管闭锁伴半椎体患者携带一个GLI3的新生突变(M111T),进一步行蛋白功能实验发现:该突变可导致内源性SKIL表达下降,并且GLI3与SKI家族蛋白-SKIL间的相互关系减弱,提示该突变可通过影响GLI3与SKIL的相互作用导致患者出现食管闭锁及半椎体表型。2019年,Renard et al[14]首次发现一个GLI3基因SNP(c.4609C>T,rs35364414)在人类腰骶部NTDs中富集。研究显示,人类腰骶部NTDs患者中存在GLI3的新生突变(H194Y),该突变同处于GLI3基因的Repressor域,该区域主要参与同SKI家族蛋白的相互作用,且GLI3基因M111T突变患者表型中存在半椎体,因此考虑GLI3基因的新生突变(H194Y)可能为该家系中患者的发病原因。后续仍需对H194Y突变进行功能试验进一步验证。

图4 Q100家系成员GLI3基因变异Sanger测序验证结果S:患者;M:母亲;F:父亲
3 讨论
NTDs作为多基因疾病,存在高度遗传异质性。绝大多数NTDs病因不明。纤毛在生物的胚胎发生和维持器官完整性中发挥重要作用。纤毛的功能异常会造成一系列表型复杂且严重的人类疾病称为“纤毛病”。NTDs是一系列纤毛病的重要表型之一,如MKS患儿有脑膨出、多囊肾及并趾(指)等表型[8]。同时NTD小鼠也存在纤毛异常表型[3]。有研究[9]报道纤毛相关基因DNAAF1基因变异与人类NTDs发生相关。本课题组前期研究[10]显示,纤毛基因的拷贝数变异与人类NTDs相关。
本研究利用AmpliSeq技术设计了包括纤毛极性基因(如PARD3)及明确纤毛病候选基因在内的49个相关基因的编码区靶向捕获Panel,对56个散发腰骶部NTDs家系进行测序,发现两个可能与神经管畸形表型发生相关的基因:CRB2及GLI3。
CRB2与CRB1及CRB3为同源基因,均为Crumbs蛋白家族成员,在视网膜、大脑和肾脏中均有表达。Crumbs蛋白复合物(CRB-PALS1-PATJ或CRB-PALS1-MUPP1)与PAR蛋白复合物(PAR3-PAR6-aPKC)对发育的神经上皮细胞建立细胞极性有重要作用[11]。Boroviak et al[12]对CRB2基因的功能研究表明CRB2对维持其他极性细胞的稳定性也是必要的,无CRB2基因表达的胚胎干细胞在神经细胞开始分化时无法存活。人类CRB2基因突变可导致局灶性肾小球硬化(FGSG-9,MIM:616220)及一些纤毛相关疾病。本研究首次在NTDs家系发现CRB2基因突变,虽然其对神经管畸形的致病性尚不明确,但课题组前期通过病例对照研究[6]发现与CRB2基因密切作用的复合物——PARD3基因突变是人类NTDs的危险因素;通过突变子的体外细胞功能实验和基因敲除的鸡胚形态学实验证实,PARD3基因aPKC结构域突变后可导致神经上皮细胞极性消失,该基因敲除后神经管腔化异常。小鼠模型研究[13]发现,CRB2基因敲除后,PALS1和PARD3基因表达明显下调,而PALS1基因敲除后MDCK细胞的管腔化异常。本研究中1例单纯腰骶部神经管畸形家系患者携带CRB2的复杂杂合变异,软件预测均为危害性变异,考虑可能为该家系中患者的发病原因。尚需通过更多的病例-对照研究进一步证实CRB2基因罕见变异和人NTDs发生的关系。
