一家系遗传性多发性骨软骨瘤的致病突变基因鉴定
2022-05-05张砚卓李闪吴成爱陶剑锋王倩倩于淑男魏祯杰姜旭
张砚卓,李闪,吴成爱,陶剑锋,王倩倩,于淑男,魏祯杰,姜旭
1 北京市创伤骨科研究所分子骨科研究室,北京100035;2 北京积水潭医院矫形骨科
遗传性多发性骨软骨瘤(HME)又称遗传性多发性外生骨疣,是一种罕见的常染色体显性遗传的骨骼肌肉疾病,发病率约为1/50 000[1]。HME 的外显率100%,主要特征是长骨干骺端外生多处软骨骨疣,70%~90%的受累部位发生在股骨远端、胫骨近端、腓骨和肱骨[2],常伴有慢性疼痛、关节活动受限、肢体畸形、身材矮小、脊柱侧凸和神经血管异常等症状。2%的HME 患者骨软骨瘤会发生恶化,转变为骨癌或骨肉瘤[3]。HME 具有遗传异质性,目前已报道3 个致病基因,分别为EXT1(MIM 608177)、EXT2(MIM 608210)和EXT3(MIM 600209)[4]。70%~95%的HME 患者由EXT1或EXT2基因突变导致,这两同源基因编码高尔基相关糖基转移酶,参与硫酸肝素生物合成[5-6]。HME 患者后代患病风险高达50%,具有较高的遗传性和致畸性,因此有必要进行基因检测或产前诊断[7]。2018年9月—2021年5月,我们收集一个中国汉族人HME 家系,对该家系中5例患者进行致病突变鉴定,以明确其遗传学病因,为家系的遗传咨询和产前诊断提供参考依据。
1 资料与方法
1.1 临床资料 入选对象为2018 年就诊于北京积水潭医院的一个HME家系。先证者(Ⅲ3)出生时未见异常,因膝关节活动受限、疼痛2018 年到我院矫形骨科就诊。经X线检查胸骨、肋骨、肩胛骨、股骨、胫腓骨、手指、脚趾头等共有20 处软骨瘤(图1a~d)。对先证者家系成员进行X线检查,明确其父亲、姐姐和姐姐的两个孩子均患病。该HME 家系图见图2。先证者(Ⅲ3)自述其2岁时发病。他的父亲Ⅱ2自述7岁时发病,在股骨、胫腓骨、手指、前臂、脚踝共有6处软骨瘤(图1e)。先证者的姐姐Ⅲ2,发病年龄1 岁,脚底板、手指、胫骨共有6 处软骨瘤(图1f)。先证者大外甥Ⅳ1,发病年龄7 个月,在肋骨、肩胛骨、前臂、手指、胫骨、腓骨、脚踝共有16 处软骨瘤(图1g)。先证者小外甥IV2,发病年龄7 个月,在肋骨、肱骨、股骨、胫腓骨、锁骨、前臂、手指共有22 处软骨瘤(图1h)。患者受累部位主要集中在股骨和胫骨。所有患者因骨疣存在,使时常活动受限,并且患者父亲和其俩外甥均有肢体畸形的表型,身高也较正常人偏矮。先证者(Ⅲ3)的母亲(Ⅱ7)无任何多发性骨软骨瘤的临床表现,血液生化学检测发现:先证者、先证者之父(Ⅱ3)、姐姐(Ⅲ2)和两个外甥(Ⅳ1和Ⅳ2)血清高密度脂蛋白胆固醇(HDL-C)水平分别为1.05、1.09、1.07、1.07、1.08 mmol/L 低于正常值(1.16~1.42 mmol/L)。在取得家系成员的知情同意后,采集家系中5 名患者和II7 静脉外周血4~5 mL。EDTA抗凝,置于4 ℃保存。本研究获得北京积水潭医院伦理委员会批准,家系成员均签署知情同意书。
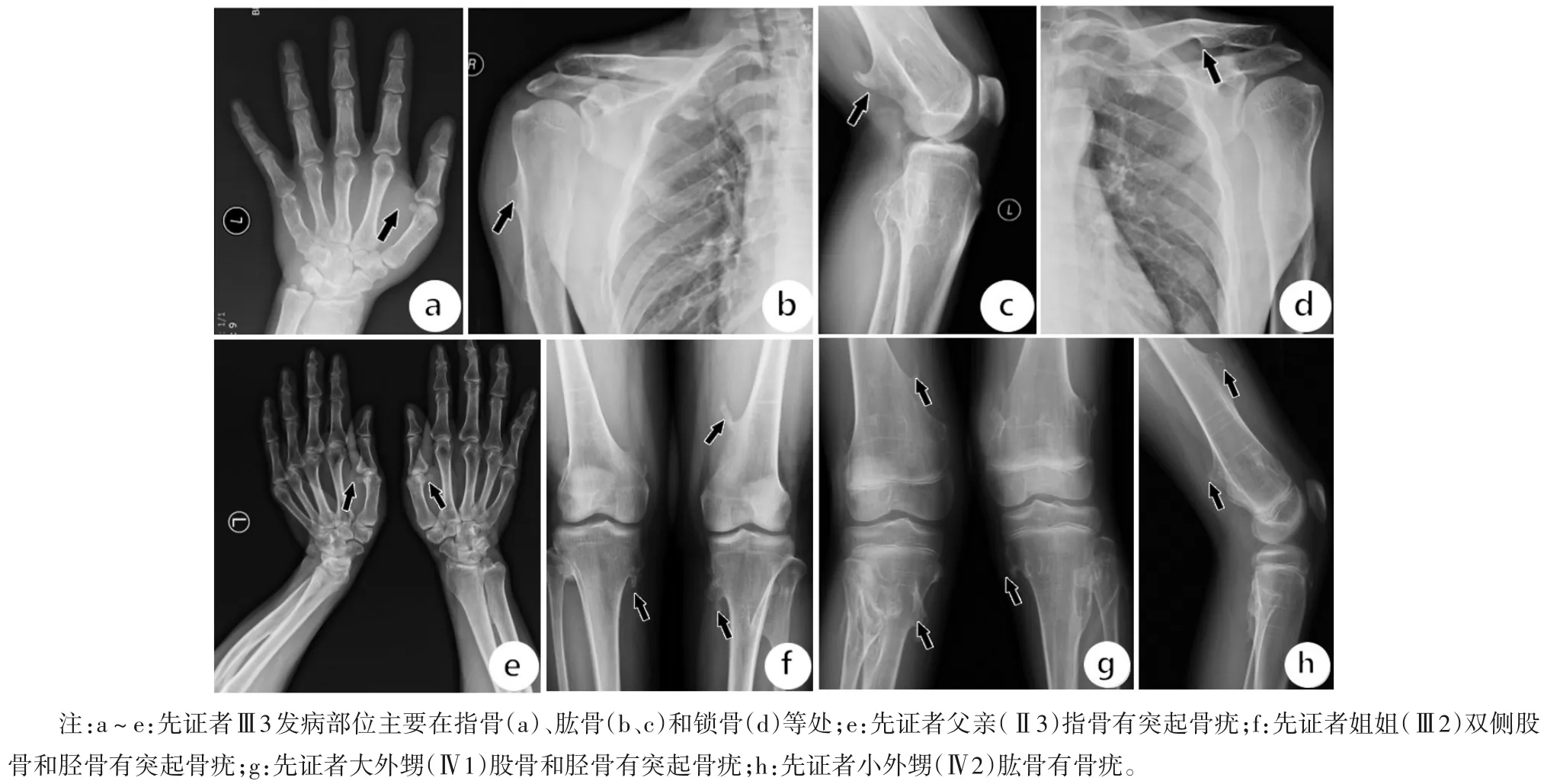
图1 HME家系X线影像图
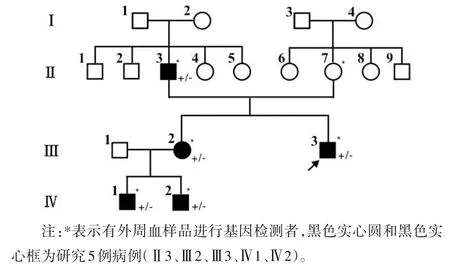
图2 HME家系图
1.2 HME家系致病突变基因鉴定
1.2.1 基因组DNA提取 采用QIAamp(Qiagen:货号51104)血液基因组小量提取试剂盒提取外周血基因组DNA,-20 ℃保存备用。
1.2.2 全外显子组测序 本研究的全外显子组测序,委托北京诺禾致源科技股份有限公司完成。将基因组DNA经Covaris破碎仪随机打断成180~280 bp片段,经末端修复和加A尾后在片段两端分别连接上接头制备DNA 文库。带有特异index 的文库pooling后与生物素标记的探针进行液相杂交,再使用带链霉素的磁珠将基因上的外显子捕获下来,经PCR 扩增后进行文库质检,合格即可进行测序。文库构建完成后,先使用Qubit 2.0 进行初步定量,随后使用NGS3K/Caliper 对文库的insert size 进行检测,insert size 符合预期后,使用qPCR 方法对文库的有效浓度(3 nmol/L)进行准确定量,以保证文库质量。库检合格,运用Illumina HiSeq2500平台进行测序。
1.2.3 生物信息学分析 测序得到的数据经过基本数据过滤(Base calling、过滤接头序列、去污染),运用Burrows-Wheeler Aligner(BWA)软件比对至人类基因组参考序列GRCh37/hg19 上,经ANNOVAR软件对变异位点进行注释,筛选致病变异。应用千人基因组数据(1000 g_all)、ExAC 数据库、ESP6500数据库(esp6500si_all)、gnomAD 数据(gnomAD_ALL和gnomAD_EAS)和dsSNP 等频率数据库,过滤掉频率高于1%的突变。应用预测软件SIFT(http://sift.jcvi.org/)、Polyphen-2(http://genetics.bwh.harvard.edu/pph2/)、PROVEAN(http://provean.jcvi.org/seq_submit.php)、Mutation Taster(http://www.mutationtaster.org)、M-CAP(http://bejerano.stanford.edu/mcap/)和CADD(http://cadd.gs.washington.edu/)对变异位点进行致病性预测。同时使用数据库ClinVar(https://www.ncbi.nlm.nih.gov/clinvar/)、OMIM(https://omim.org)和HGMD(http://www.hgmd.cf.ac.uk/ac/index.php)确定变异是否被收录。根据2015年美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics,ACMG)指南对变异致病性进行分级。
1.2.4 PCR-Sanger 测序 从UCSC 人类基因组数据库获得EXT1(NM_000127.2)基因序列,使用在线引物设计软件Primer5.0,针对候选变异设计特异性引 物(EXT1-E6-F:5’-ATACCATGGGAGAGACAGCA-3’和EXT1-E6-R:5’-GTAACGAGGCAGGATGAATG-3’),以家系成员基因组DNA 为模板,扩增变异位点所在的EXT1基因的第六个外显子。PCR反应体系:正反向引物各0.75 μL(10 pmol/μL),La taq 聚合酶0.25 μL,2×GC buffer I 12.5 μL,无菌去离子水6.25 μL,DNA 模板100 ng;PCR 反应条件:95 ℃预变性3 min;94 ℃变性30 s,58 ℃退火30 s,72 ℃延伸30 s,进行36 个循环;72 ℃延伸8 min。测序产物送至北京诺赛基因组研究中心有限公司,采用ABI3730xl 测序仪进行测序。测序结果经Codon-Code Aligner序列分析软件分析。
2 结果
根据全外显子组测序结果,用Sanger 测序对先证者和家人进行突变验证。结果表明:先证者EXT1基因第6 外显子存在杂合突变c.1468dupC,该突变导致EXT1基因编码的蛋白第490 位亮氨酸转变为脯氨酸,并在其后的第31 位形成终止密码子(p.Leu490Profs*31,见图3);该突变可引起EXT1基因mRNA 的翻译提前终止,导致蛋白质功能缺失。家系中其他4例患者(Ⅱ3、Ⅲ2、Ⅳ1和Ⅳ2)均检测到与先证者相同的突变(c.1468dupC),表型正常的家系成员未检测到该位点的变异。家系验证结果表明,家系内存在基因型与表型共分离。多个同源序列比对分析表明,EXT1基因c.1468编码的第490位亮氨酸在多种不同生物种属内均是保守的。
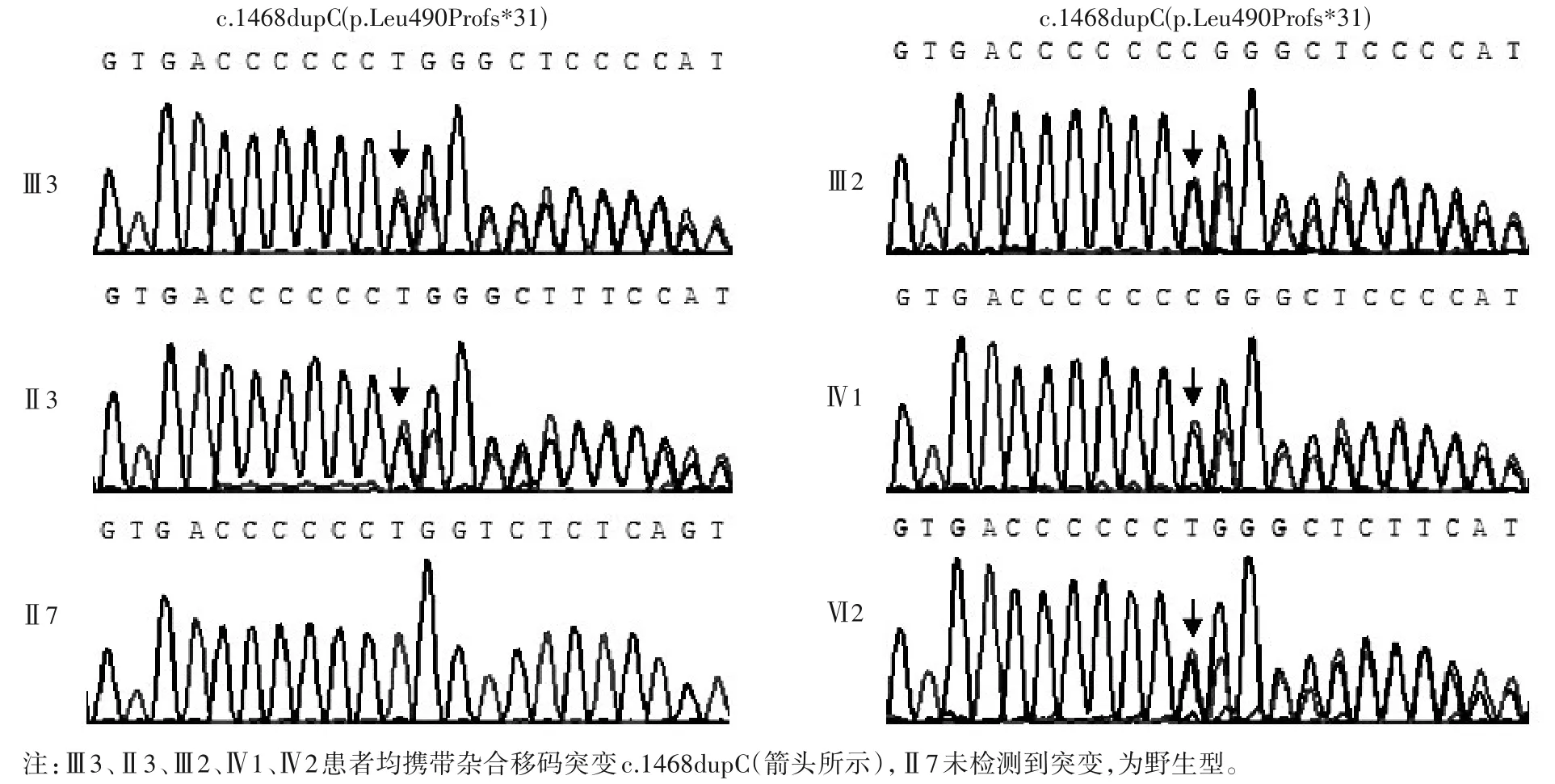
图3 EXT1基因c.1468dupC变异位点Sanger测序结果
3 讨论
HME 是一种遗传异质性的骨骼系统疾病,患病率为1/50 000,主要影响软骨内骨生长。外生骨疣的数量和大小不一,病变的位置也不同[8-9]。该疾病的特征是形成大量的软骨帽和良性骨肿瘤(外生骨软骨或骨软骨瘤),通常伴有骨骼畸形和身材矮小。在少数患者中,外生软骨会发生恶性转化,导致骨肉瘤或软骨肉瘤[10]。HME 患者膝关节畸形的发生率很高,约有1/3 的患者发展为膝外翻。30%~60%的HME患者中可见前臂弓状畸形,患者其他骨骼畸形还包括肢体长度不一致、下肢外翻畸形和脊柱侧凸。与女性患者相比,男性患者通常表现出更严重的症状,这可能是由于男性生长板闭合较晚,从而有更多的时间形成外生骨瘤造成的。此外,有较多外生骨的患者通常有较严重的残疾和畸形[10]。该疾病没有特异性药物,只能在恶变的情况下手术切除症状最明显、问题最严重的骨软骨瘤[3]。70%~95%的HME 患者是由EXT1和EXT2突变所致,其中EXT1占56%~78%,EXT2占21%~44%[10]。EXT基因家族由肿瘤抑制基因组成,其功能的丧失或失调导致HME。EXT蛋白是定位于内质网的Ⅱ型跨膜蛋白,包括一个N 端细胞质尾部、一个跨膜结构域、一个柄和一个大的球状结构域[11-13]。EXT1和EXT2在高尔基体中形成异寡聚糖基转移酶,并与糖基转移酶活性密切相关,通过交替添加GlcAc 和GlcNAc残基延长HS链,参与硫酸肝素(HS)聚合[14]。EXT1/EXT2复合体比单独的EXT1或EXT2具有更高的糖基转移酶活性。由于HME 具有异质性,且EXT1/EXT2基因片段较大,又缺少突变热点,应用WES 技术于HME患者突变鉴定[15]。
多发性骨软骨瘤通常在儿童和青少年时期增加骨肉瘤的大小和数量。尽管它们对长骨的近骨骺端区有偏好,但事实上可以出现在身体的任何骨骼上,包括短骨、扁骨和不规则骨[12]。研究[16-18]表明,许多HME 患者在发病早期甚至后期都没有任何临床症状。在我院矫形骨科接诊的患者中,一家系中三代出现患者,且全部自愿接受致病突变基因鉴定的情况并不常见。本研究所收集的家系中,所有患者因外生骨疣使活动受限,但软骨未恶化成骨肉瘤或软骨肉瘤,且患者未采用手术治疗。家系中成年患者(Ⅱ2、Ⅲ2 和Ⅲ3)身高均低于正常人,且先证者父亲(Ⅱ2)和他俩外甥(Ⅳ1 和Ⅳ2)均有四肢畸形。从患者的临床表型、四肢畸形和骨疣数量,我们观察到患者严重程度在家系中呈现出早现的现象。在患者血液生化检测中发现,HDL-C 低于正常值。HDL-C 是体内重要的脂蛋白,它的生物学功能主要体现在参与胆固醇的逆向转运,改善血管内皮功能,减少黏附因子表达,逆转低密度脂蛋白氧化及抗凋亡等多个方面。研究发现HDL-C 水平与心血管病风险呈负相关,血浆HDL-C 每上升0.026 mmol/L,心血管事件发生风险可下降2%~3%。在血浆HDL-C组成及形成过程中最关键蛋白为apoA1和ABCA1[17]。在我们的研究中未发现apoA1 和ABCA1 的异常(含量及基因检测)。至今,未见HME 与HDL-C 相关性研究报道。HME 导致血浆HDL-C 水平降低还是两者互不相干而是由其他原因引起的HDL-C 水平降低,这些问题我们将进一步进行探究。
本研究我们通过全外显子组测序对一个常染色体显性遗传的HME家系进行致病突变分析,发现该家系是由EXT1基因第6 外显子的一个移码突变c.1468dupC(p.Leu490Profs*31)导致,该突变导致第490位后的编码氨基酸发生移码,并在第521位引入终止密码,使EXT1基因编码全长为746氨基酸组成的肽链缩短至由521 个氨基酸组成的截断型蛋白。EXT1基因编码硫酸肝素生物合成所需的糖基转移酶。EXT1基因上的致病突变引起硫酸乙酰肝素聚合酶合成障碍或降解,调节软骨细胞增殖成熟的信号被破坏,导致骨骺生长板发育异常,但其中的具体调控机制并不清楚[11-15]。迄今为止,数据库Human Gene Mutation Database (HGMD,http://www.hgmd.cf.ac.uk/ac/search.html)和Multiple Osteochondromas Mutation Database(MOdb,http://medgen.ua.ac.be/LOVD)已报道数563 种EXT1和267种EXT2基因突变与HME 相关,随机分布于各个外显子区域[6]。大多数已知的致病变异会导致截断蛋白的产生,如移码突变、无义突变和剪接位点突变,错义变异并不常见。检索HGMD 数据库发现,本研究在一个中国人HME家系中发现的上述突变,曾在日本人群中被报道,再次证实了该突变的致病性[16]。
总之,我们在中国HME 家系中发现一个EXT1基因的突变,该结果进一步扩展了EXT1基因的突变谱,有利于后续的遗传咨询和产前诊断,也为研究HME发病机制提供更多依据。
