HCN气体在金属Cu、Zn表面吸附的密度泛函研究
2021-12-27张艳琨杨春晓张可欣李粉吉吕振东夏福婷彭金辉
张艳琨, 杨春晓, 张可欣, 李粉吉, 吕振东, 夏福婷, , 彭金辉
(1.云南民族大学 化学与环境学院 云南省高校民族地区资源清洁转化重点实验室, 昆明 650500; 2.昆明理工大学 冶金与能源工程学院 复合有色金属资源清洁利用国家重点实验室, 昆明 650093)
1 前 言
氰化氢(HCN)是一种典型的“非常规”有毒有害气体污染物,广泛存在于煤化工、石油炼制、冶金、磷化工、合成氨等工业废气和机动车尾气中[1]. 它能够抑制人体呼吸酶,引起细胞内窒息,对人体极为有害[2, 3]. 人在含量小于20 ppm HCN的环境中即可感到胸部压迫感而缓慢中毒致死,当含量上升为300 ppm时暴露1分钟即会迅速致命[4]. 去除气相中HCN的方法主要有吸附法、催化氧化法、水解-氧化耦合法等. HCN与金属之间的吸附在催化反应中起着重要作用[5]. 研究表明HCN可以在过渡金属表面发生解离生成C2N2和H2等物种[6-8],因此,研究HCN在过渡金属表面的吸附对HCN的消除净化有很强的指导意义.
Cu和Zn因其特有的d电子结构,使他们具有良好的活性、稳定性和选择性,因而成为重要的过渡金属催化剂. Pd、Pt、Cu等作为活性组分的催化剂具有较高的HCN催化水解活性,具有降低SCR脱硝反应中HCN含量的潜力[9]. 有研究证明,随着催化剂上Cu负载量的增加,低温段HCN的转化率和N2选择性均有所提高[10],催化剂中Zn和Ni相对含量的多少对催化剂催化水解HCN的活性有较大的影响[11]. 以γ-Al2O3为载体制备的HCN的水解催化剂负载各种过渡金属盐类时,其水解催化剂活性有所增加[12]. Bridge课题组[13]采用LEED、AES、程序脱附光谱(TDS)以及热脱附质谱仪(TDMS)研究了HCN在Pt(110)、Ag(110)表面上的吸附情况,认为HCN能够分解生成CNad物种. 然而HCN的剧毒性带来的实验过程高风险、实验条件苛刻和高运行成本限制了研究人员对其进行深入的实验研究. 密度泛函理论是从电子结构出发借助基本常量和合理近似进行的计算方法,它可以从电子和原子层面理解吸附过程. Celio等[14]推测HCN是通过N原子垂直吸附在于Pt(111)和Cu(100)表面上的. 胡建明等[15]在DFT框架下,采用簇模型方法,对包括吸附键在内的所有键长进行优化,并在此基础上优化了吸附键角,研究了HCN和HNC分子在Cu(100)面的吸附解离状况,探讨了HCN和HNC分子在Cu催化剂表面上不同吸附位的稳定性、吸附能以及成键情况. 本文采用密度泛函理论方法进一步研究了HCN在Cu(100)、Cu(110)、Cu(111)、Zn(100)、Zn(110)、Zn(111)表面上不同吸附位点的吸附性质. 通过分析对比吸附能,电子态密度以及电荷转移情况,得到HCN在Zn、Cu不同表面上最稳定的吸附构型. 本文的研究对于Cu、Zn催化机理的研究和金属催化剂性能的改进具有重要意义.
2 计算方法及模型
本文中所有的理论计算均采用Materials-Studio(MS)软件包中的DMol3软件包完成. 采用广义梯度近似(GGA)中的Perdew-Burke-Ernzerh(PBE)交换关联势描述电子交换相关作用. 构建2×2的超晶胞,为了避免各平板间的相互作用将真空层设为15 Å,同时,固定最底层原子[16]. 采用2×2×1的K点进行结构优化和能量计算. 原子结构优化中的能量收敛性判据为1.0-6Ha,每个原子受力不大于0.002 Ha/ Å,最大位移为0.005 Å. 在全部的计算过程smearing参数设置为0.008 Ha,优化几何结构时,基组截断设置为3.5. 计算精度为良好. HCN在金属衬底吸附时,吸附能的计算公式为[16]:Eads=EHCN/slab-(Eslab+EHCN).Eads为吸附能,EHCN/slab为HCN吸附在表面时体系的总能量,Eslab为表面的能量,EHCN为吸附前HCN的能量. 吸附是放热反应,吸附能计算值为负值表示吸附后体系的稳定性增强,且吸附值越小体系的稳定性越强[17].
3 结果与讨论
3.1 理论模型
优化后铜的晶胞参数a=b=c=3.61 Å,α=β=γ=90°,锌的晶胞参数a=b=2.66 Å,c=4.95 Å,α=β=90°,γ=120°,与实验值都是相符的,说明计算方法是合理的. 图1A、B、C是不同的Cu表面,每个表面做了三个吸附位点. 图1D、E、F是不同的Zn表面,两个表面做了三个吸附位点,其中Zn(111)表面做了两个桥式吸附位点. 由于HCN易于通过具有孤对电子的N原子垂直吸附在金属表面上[16]因此Top为垂直的顶式吸附位,N原子垂直吸附在Cu(Zn)原子上,Bridge为垂直的桥式吸附位,N原子垂直作用在Cu-Cu(Zn-Zn)键上. Hollow为中空吸附位N原子和C原子分别作用在两边的Cu(Zn)原子上.

图1 Cu,Zn不同表面的吸附位点的俯视图Fig. 1 Top views of adsorption sites on different surfaces of Cu, Zn
3.2 HCN的吸附性质
对于气相HCN分子,计算的C-H键长为1.076 Å,C≡N键长为1.163 Å,HCN的键角为179.9°,与实验值[18]1.064 Å,1.156 Å和180°相差不大. 表1为HCN在不同的Cu、Zn表面上吸附后HCN的结构参数以及吸附能量. 由表1可看出,在HCN/Cu(100)体系中,各吸附位点的稳定性排序为Bridge>Top=Hollow. Bridge吸附位点的吸附能为-0.42 eV,属于物理吸附[19]. HCN吸附在Cu(110)表面时,各吸附位点的稳定性排序为Top=Hollow>Bridge. Top、Hollow吸附位点的吸附能都为-0.42 eV,属于物理吸附. 在Hollow吸附位点上,HCN中的C≡N键由原始的1.163 Å变为1.167 Å,被拉长了0.004 Å,C≡N被拉长说明该位点对HCN的催化活性强,该位点的键角变化率为0.83%,较大的键角变化率说明表面对HCN有较强的吸附能力[16]. HCN吸附在Cu(111)表面时,三个吸附位点的稳定顺序为Top>Hollow=Bridge,都属于物理吸附. 对比吸附能数值可知,HCN在不同Cu表面的所有吸附位点上,最稳定的吸附位点为Cu(110)表面的Top吸附位点.
当HCN吸附在Zn(100)表面上时,各吸附位点的稳定性情况排序为Hollow>Bridge>Top,吸附能都是负值,说明HCN吸附在该位点上都是放热反应,属于物理吸附. 对于HCN/Zn(110)体系,Bridge吸附位点为最稳定吸附位点,属于物理吸附,其次是Hollow吸附位点,Top吸附位点. Zn(111)表面对HCN的吸附能力比较强,其吸附稳定性排序为Hollow>Top>Bridge2>Bridge. 其吸附能数值均较大,属于化学吸附. 在Hollow吸附位点上,HCN中的C≡N键由原始的1.163 Å变为1.165 Å,被拉长了0.002 Å,比在该表面的其他吸附位点上的变化都大,该位点的键角变化率为0.67%,其变化率大于Zn表面的其他吸附位点,根据前人的研究经验,HCN的键角变化率越大,表面对HCN的吸附能力越强[16]. 从HCN在不同Cu、Zn表面的吸附能数值来看,金属Zn对HCN的吸附能力更强.

表1 计算得到的HCN在不同Cu,Zn表面上的吸附能和吸附结构参数
3.3 Mulliken电荷布居分析
为了更准确地了解HCN与Cu、Zn不同表面的相互作用机制,对吸附前后的HCN进行Mulliken电荷布局分析. 表2是HCN中C、H、N原子和Cu、Zn不同表面之间电荷转移的变化(ΔQ)和转移数量的总和. 正值ΔQ意味着失去电子,负值意味着获得电子[20]. 电荷转移值为正值时表示电荷从HCN转移到表面,为负值时表示电荷从表面转移到HCN. 当HCN吸附在Cu表面时,电荷转移值均为正值,电荷从HCN转移到Cu表面. 在HCN/Cu(110)体系中,Bridge吸附位点电荷转移值为0.220 e,Top吸附位点的电荷转移值为0.222 e, Hollow吸附位点电荷转移值为0.223 e,这三个吸附位点的吸附能相差不大,其电荷转移值也相差不多,Cu(100)、Cu(111)表面也是同样的规律. 当HCN吸附在Zn表面上时,Zn(110)表面的Bridge吸附位点的电荷转移值为-0.007 e,Hollow吸附位点的电荷转移值为-0.055 e,并且在该位点上,HCN中的C原子失去电子,在Zn(110)表面的三个吸附位点中只有该位点的C原子失去电子,因此该吸附位点的电荷转移值比较大. 在Zn(100)表面的Bridge吸附位点的电荷转移为-0.005 e,Hollow吸附位点的电荷转移为-0.016 e,HCN中的C、N原子在这两个吸附位点上均失去电子,一部分转移到HCN分子中的H原子上,一部分转移到Zn表面,以增强表面的吸附能力. 在Zn(111)表面中,Bridge2、Hollow吸附位点的电荷转移值均为负值,说明电荷从Zn表面转移到HCN以增强Zn表面的吸附能力[21],Hollow吸附位点的电荷转移数量较多,说明在该位点上Zn(111)表面对HCN的吸附能力更强,这与分析体系吸附能分析结果一致.

表2 HCN在不同Cu,Zn表面吸附后电荷转移的变化
3.4 态密度分析
利用Dmol3模块的Analysis功能可以对优化后的模型进行电子态密度的分析. 根据同一表面不同位点吸附前后HCN的态密度变化的分析,可以进一步理解吸附能差异的原因,而HCN与Cu、Zn不同表面最稳定吸附前后电子态密度变化可以深入解释表面对HCN的吸附强弱. 图2A为HCN吸附前的态密度图,图2B为HCN吸附在Cu(100)表面后的态密度图,图2C为HCN吸附在Zn(100)表面后的态密度图. 由图2B、C和图2A对比可知,吸附后的HCN的态密度向低能级移动,说明HCN在Cu(100)、Zn(100)表面吸附后能量降低,比气相中HCN的结构更加稳定. HCN吸附在Cu(100)表面的三个吸附位点的态密度几乎重合,表明HCN在这三个吸附位点上稳定性变化范围不大,与分析吸附能所得结论一致[22]. HCN吸附在Zn(100)表面的三个吸附位点的态密度相差较大,Hollow吸附位点在1.63~3.27 eV区间态密度的峰值明显比其它两个吸附位点都要低,表明HCN吸附在该位点的能量下降最大,吸附结构较其它两个吸附位点更加稳定[23],与吸附能为负值且最小相对应.
图3A为HCN在Cu(110)表面最稳定吸附位点、最不稳定的吸附位点与吸附前Cu(110)表面的态密度. Cu(110)表面的Hollow吸附位点、Bridge吸附位点的态密度几乎重合,且都与吸附前的Cu(110)表面的态密度基本重合,两个吸附位点的态密度的范围更宽,在-5~0 eV区间内,态密度峰值较吸附前Cu(110)表面的要低,电子向低能级发生转移,体系较吸附前更加稳定. 分析之前吸附能结论可知,HCN在Cu(110)的Hollow、Bridge吸附位点的吸附值均为负值,且吸附能的值相近. 由此可知,分析吸附能所得结论和态密度分析结果一致. 图 3B为HCN在Zn(111)表面最稳定吸附位点、最不稳定的吸附位点与吸附前Zn(111)表面的态密度. 两个吸附位点的态密度与吸附前的Zn(111)表面的态密度有所差异,特别是Hollow吸附位点的态密度整体向低能级移动,在-10~-5 eV区间内,该位点态密度峰值下降较多,比Bridge吸附位点的态密度峰值下降明显,说明Hollow吸附位点能量降低的最多,结构最稳定,与之前吸附能分析结论相一致.
4 结 论
基于密度泛函理论方法研究了HCN在Cu(100)、Cu(110)、Cu(111)、Zn(100)、Zn(110)、Zn(111)表面上不同吸附位点的不同吸附情况,计算并分析了吸附能,电子态密度以及电荷转移值. HCN结构参数分析表明:在同一吸附表面的不同吸附位点上,HCN吸附后键长键角的变化越大,说明吸附作用对HCN的影响越大. 电荷分析表明,电子从吸附剂表面转移到吸附质时,可以增强吸附剂对吸附质的吸附能力,使吸附结构变得更稳定. 电子态密度分析表明,金属表面吸附HCN后,HCN的态密度整体向低能级移动,且峰值降低,吸附结构变得更稳定. 通过对比吸附能可知,HCN在Cu(100)、Cu(110)、Cu(111)三个面的吸附状态相差不大,HCN在Zn表面的吸附要强于在Cu表面的吸附.
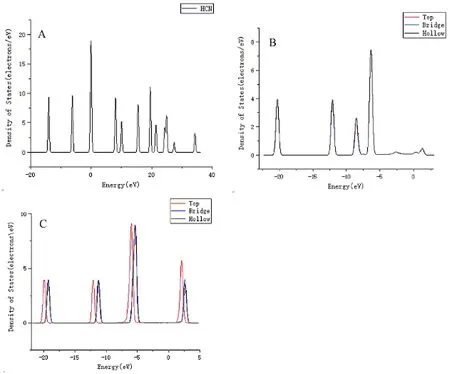
图2 HCN吸附前后态密度Fig. 2 DOSs of HCN before and after adsorption

图3 Cu(110),Zn(111)面吸附前后态密度Fig. 3 DOSs of Cu(110), Zn(111) surface before and after adsorption
