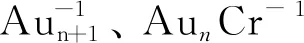团簇吸附CO的密度泛函理论研究
2021-12-27王必利
王必利, 宋 蕊, 仝 亮
(陆军工程大学 基础部, 南京 211101)
1 引 言
团簇是由几个乃至上千个原子、 分子或离子通过物理或化学结合力组成的相对稳定的微观或亚微观聚集体, 其物理和化学性质随所含的原子数目而变化, 其在表面吸附、 化学催化等方面有着十分重要的应用[1-10]. 团簇催化剂已成为当前团簇科学研究中一个相当重要的方向, 新的团簇催化材料的出现, 将对人们的生产、 生活产生重要的影响. 在团簇催化反应中, 分子在团簇表面的吸附是整个反应中最关键的一个步骤. 所以, 研究分子在团簇表面的吸附对理解团簇催化反应的机理, 研制新型团簇催化材料, 寻找合适的催化剂有着重要的意义. 此外, CO是大气中分布最广和数量最多的污染物, 如何消除CO成为一个重要的环保难题, 因此, CO的吸附研究就显得尤为重要.
近年来, 随着计算科学的发展, 密度泛函理论(DFT)在研究小分子H2, O2, NO等在团簇表面吸附中的作用日益凸显[11-14]. 当前有了一些关于金团簇吸附一氧化碳分子的实验和理论研究[15-17], 但是团簇的尺寸较小, 而且多为中性团簇, 对于阴离子团簇的研究, 特别是对参杂阴离子团簇的实验与理论研究较少. 为了研究阴离子金团簇替代掺杂铬原子后对团簇吸附一氧化碳的影响, 课题组采用DFT理论系统研究了Aun+1-1、 AunCr-1(n=1-10)与CO的相互作用, 给出了Aun+1CO-1、 AunCrCO-1团簇的基态构型, 对比了团簇尺寸变化时其吸附能、 平均结合能、 二阶差分能、 能隙的变化, 分析了Aun+1CO-1、 AunCrCO-1(n=5-7)团簇的轨道电荷分布. 希望能为新型CO催化材料的设计提供理论上的参考.
2 计算方法
本文全部计算工作使用dmol3[18,19]程序包进行, 所有计算均设定为自旋非限制, 并且没有设置任何对称性限制. 在广义梯度近(GGA)中, 交换关联势采用Perdew-Wang(1991)NLDA function(PW91)方法, 价电荷设定为-1, 考虑到密度泛函理论中对重元素的计算需要考虑相对论效应, 因此核轨道的处理采用全电子相对论 (AER)计算, 选择了精度最高的双数值轨道基组+轨道极化函数(DNP)进行了结构优化, 电荷布局为Mulliken电荷分析. 结构优化的收敛标准为: 能量阈值1×10-6Hartree(1 Hartree=27.21 eV), 力场0.002 Hartree/ Å, 最大位移0.005 Å, 结构优化过程中, 对自旋和对称性均不作限制.
为验证所选方法的可靠性, 表一给出了采用不同泛函计算了Au2团簇的键长、 振动频率、 垂直电离势VIP、 CO的键长、 振动频率, 以及对应的实验结果. 比较表明本文所选用的计算方法与实验结果最为吻合.
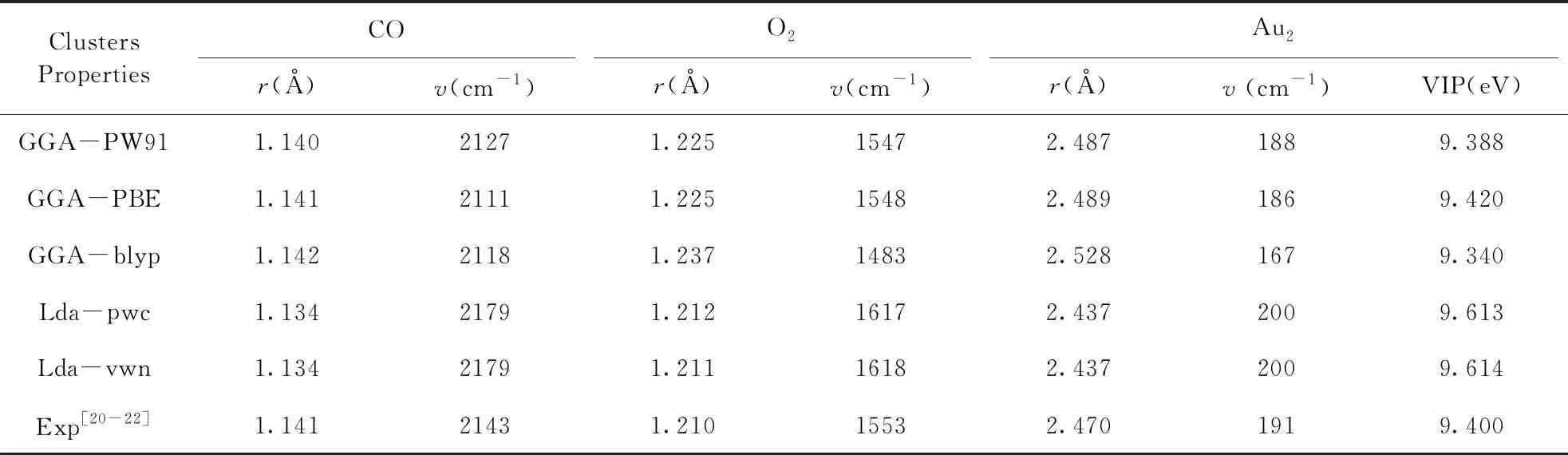
表1 不同泛函计算Au2团簇与CO多种性质的结果对比
3 结果与分析
3.1 基态结构


表2 与AunCr-1、 AunCrCO-1 (n=1-10)的基态构型(大球表示Cr原子, 未标注的小球表示Au原子)
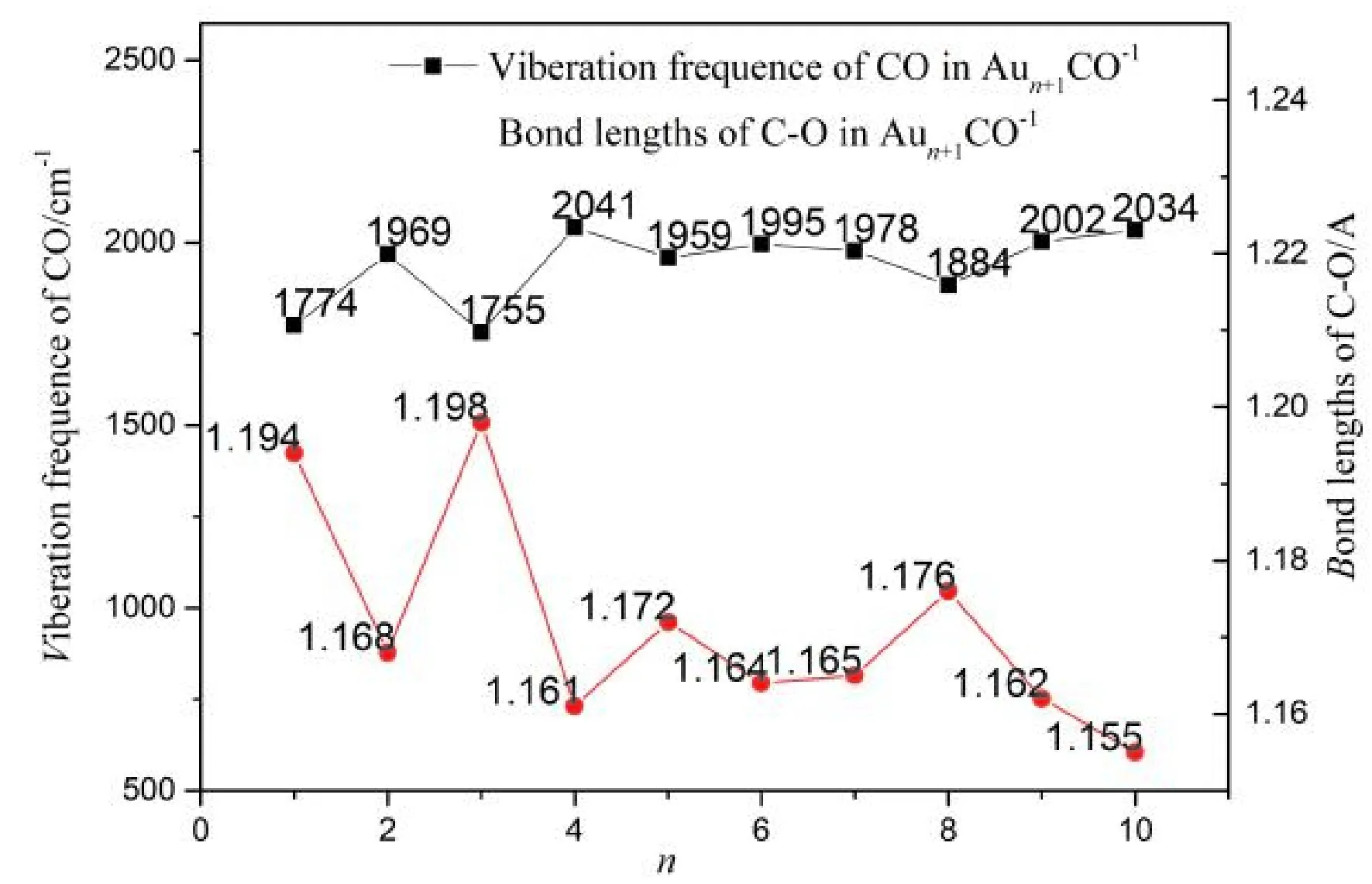
图1 Aun+1CO-1基态结构中C-O键长与CO振动频率Fig.1 Bond lengths and viberation frequencies of CO in Aun+1CO-1(n=1-10) with lowest energy structure
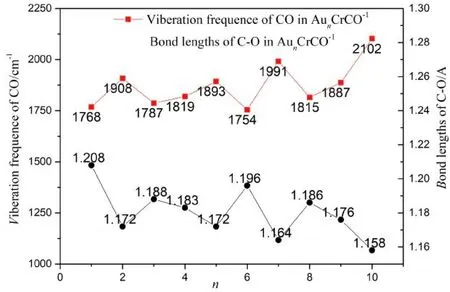
图2 AunCrCO-1 基态结构中C-O键长与CO振动频率Fig.2 Bond lengths and viberation frequencies of CO in AunCrCO-1 (n=1-10) with lowest energy structure
最后分析Aun+1CO-1、 AunCrCO-1(n=1-10)团簇中的C-O键长与CO振动频率相较气相CO分子的变化: CO分子键强减弱, 活性增强了, C-O键长越长, 表示被活化的程度越大,反之, 被活化的程度就越小; 图1与图2分别给出了Aun+1CO-1、 AunCrCO-1中C-O键长、C-O振动频率, 可以看出, Aun+1CO-1团簇中C-O键长范围在1.155 Å ~1.198 Å, 由表1可知气相CO键长为1.140 Å, C-O键长伸长量在0.015 Å ~0.058 Å, 其中Au4CO-1中C-O键长最长, 说明Au4CO-1活化最强, Au11CO-1中C-O键长最短, 说明Au4CO-1活化最弱, 总体上随着团簇尺寸的增大, C-O键长伸长量趋于减小; AunCrCO-1团簇中C-O键长范围在1.158 Å ~1.208 Å, C-O键长伸长量在0.018 Å ~0.068 Å, C-O键长变化规律与Aun+1CO-1类似. 结果表明, Aun+1CO-1、 AunCrCO-1中C-O相较气相CO分子的键长都有所增加, AunCrCO-1团簇中C-O键长相较Aun+1CO-1团簇中C-O键长长, 说明Aun+1CO-1团簇替代掺杂Cr原子,有助于CO分子的活化.
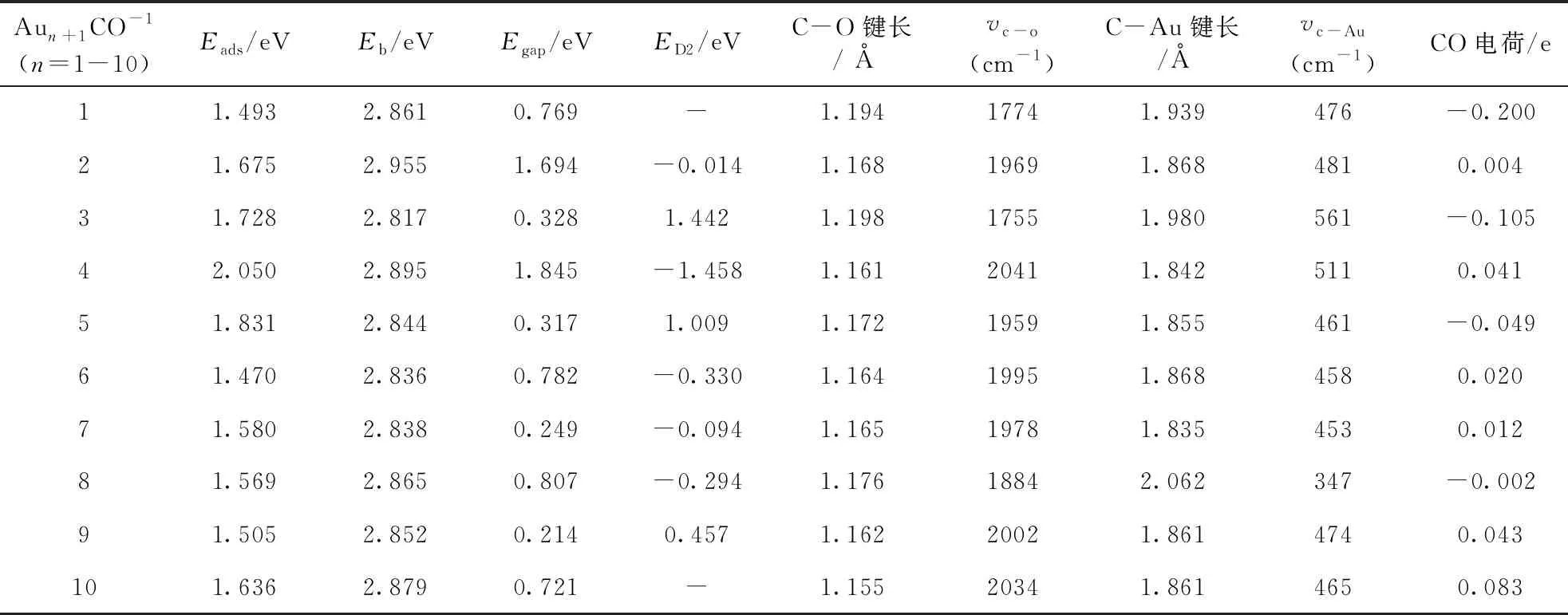
表3 团簇 Aun+1CO-1基态吸附能Eads, 结合能Eb, 能隙Egap,二阶差分能ED2, C-O键长, C-Au键长, C-O振动频率, C-Au振动频率, CO电荷
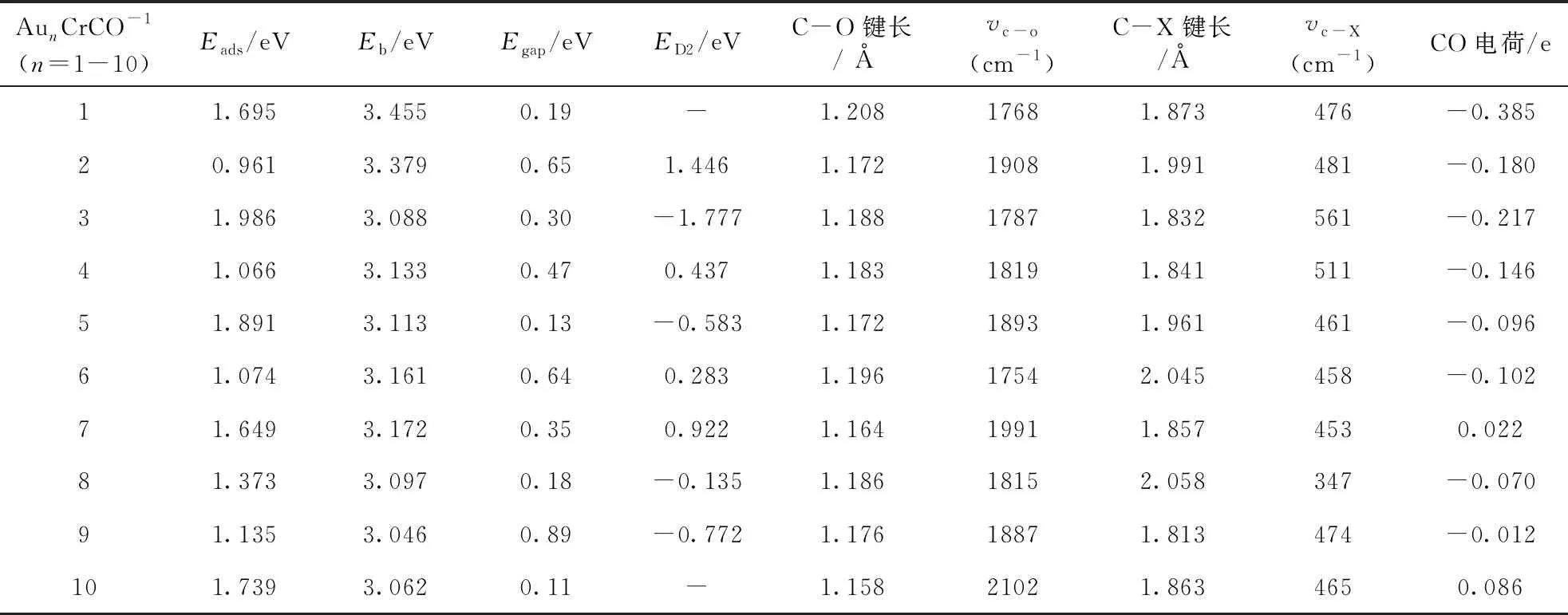
表4 团簇 AunCrCO-1基态吸附能Eads, 结合能Eb, 能隙Egap, 二阶差分能ED2,C-O键长, C-X(X=Au,Cr)键长, C-O振动频率, C-X振动频率(X=Au,Cr), CO电荷
团簇研究中, 常常用振动频率来描述成键原子间的相互作用强弱, 振动频率越高, 相互作用越强. 由图1与图2比较发现: AunCrCO-1团簇中CO振动频率总体上低于Aun+1CO-1团簇中CO振动频率, 说明AunCrCO-1团簇中C-O相互作用要弱于Aun+1CO-1团簇中C-O相互作用, 这与前面键长分析结果相吻合.
为了研究团簇替代掺杂铬原子对团簇稳定性、化学活性与吸附强度的影响, 表3、4分别给出了Aun+1CO-1、 AunCrCO-1(n=1-10)团簇的吸附能Eads、 平均结合能Eb、 能二阶差分能ED2、 隙Egap、 C-O键长、 C-Au/Cr键长、C-O振动频率、以及CO电荷. Aun+1CO-1、AunCrCO-1团簇的Eads、Eb、ED2定义式如下:
Eads[Aun+1CO-1]=E[CO]+
(1)
Eads[AunCrCO-1]=E[CO]+
E[AunCr-1]-E[Aun+1CO-1]
(2)
Eb[Aun+1CO-1]=(-E[Aun+1CO-1]+
nE[Au]+E[Au-1]+E[O]+E[C])/(n+3)
(3)
Eb[AunCrCO-1]=(-E[AunCrCO-1]+
(n-1)E[Au]+E[Au-1]+E[Cr]+
E[O]+E[C])/(n+3)
(4)
ED2[Aun+1CO-1]=E[Aun+2CO-1]+
E[AunCO-1]-2E[Aun+1CO-1]
(5)
ED2[AunCrCO-1]=E[Aun+1Cr CO-1]+
E[Aun-1Cr CO-1]-2E[AunCrCO-1]
(6)
以上E(X)(X= C、 O、 Cr、 Au、 Au-1、 AunCO-1、 Aun+1CO-1、 Aun+2CO-1、 Aun-1Cr CO-1、 AunCr CO-1、 Aun+1CO-1、 Aun+1CO-1)表示对应原子与团簇的能量.
3.2 吸附强度分析
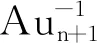
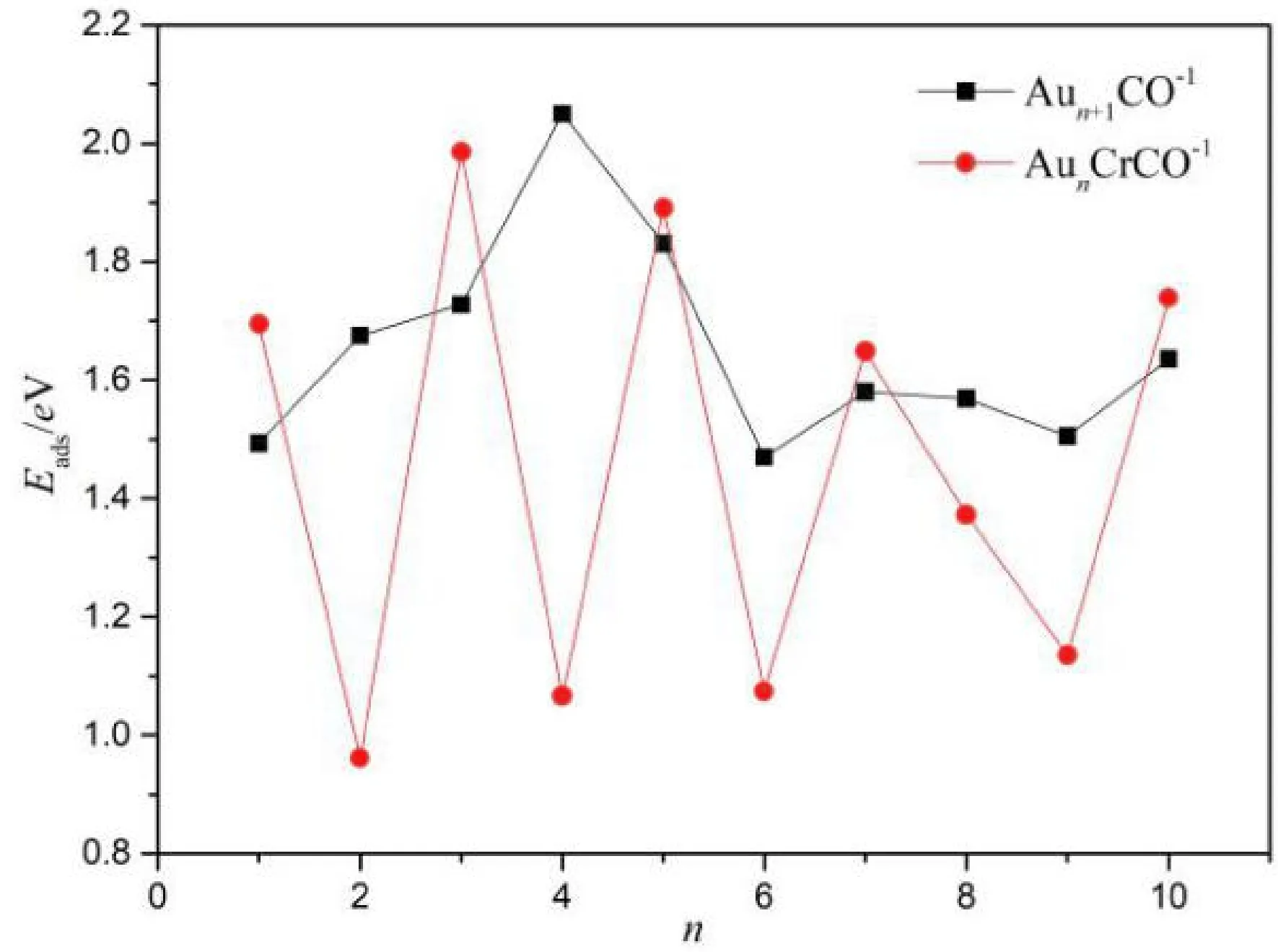
图3 Aun+1CO-1、 AunCrCO-1基态的吸附能Fig.3 Adsorption energies of Aun+1CO-1 and AunCrCO-1 (n=1-10) with lowest energy structure
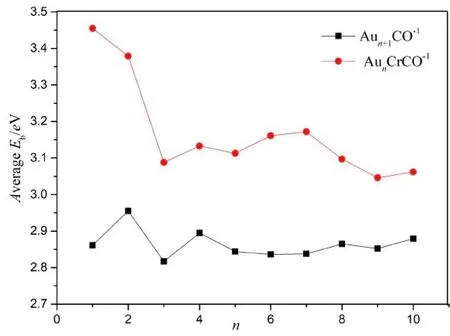
图4 Aun+1CO-1、 AunCrCO-1基态的平均结合能Fig. 4 Average binding energies of Aun+1CO-1 and AunCrCO-1 (n=1-10) with lowest energy structure
3.3 稳定性和化学活性分析
二阶差分能是描述团簇稳定性的一个很好的物理量, 其值越大, 对于团簇稳定性越大. 图5给出了Aun+1CO-1、 AunCrCO-1的二阶差分能ED2随团簇尺寸的演化规律, 可以看出:对于Aun+1CO-1团簇, 其二阶差分能具有明显的“幻数”效应, Aun+1CO-1(n=3,5,7,9)团簇的二阶差分能大于相邻的团簇, 随着团簇尺寸增大, Aun+1CO-1团簇的二阶差分能趋于稳定; 对于AunCrCO-1团簇, 其二阶差分能的不具备和Aun+1CO-1团簇一样的“奇偶”振荡性.
图5 Aun+1CO-1、 AunCrCO-1基态的二阶差分能Fig.5 Second-order difference energies of Aun+1CO-1 and AunCrCO-1 (n=1-10) with lowest energy structure
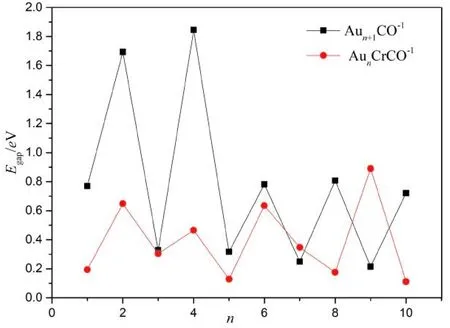
图6 Aun+1CO-1、 AunCrCO-1基态能隙Fig.6 Energy gaps of Aun+1CO-1 and AunCrCO-1 (n=1-10) with lowest energy structure
能隙反映了团簇的化学活性, 是考察物质导电性的参数, 能隙越大, 团簇化学活性越低, 反之, 能隙越小, 团簇化学活性越低. 图6给出了Aun+1CO-1、 AunCrCO-1的能隙随团簇尺寸的演化规律, 可以看出: 对于Aun+1CO-1团簇, 其能隙值与二阶差分能一样也具有“奇偶”震荡性, 偶数团簇的能隙高于相邻奇数团簇的能隙, Au5CO-1的能隙最大, 说明Au5CO-1团化学活性最低, 最不易得失电子; 对于AunCrCO-1团簇, 其能隙值随着尺寸增长而振荡, 总体上,n取值相同时AunCrCO-1的能隙小于Aun+1CO-1的能隙, 表明Aun+1CO-1替代掺杂Cr原子后团簇的化学活性得到了提升.
3.4 电荷布局分析
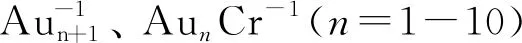

表5 Aun+1CO-1、 AunCrCO-1(n=5-7)与CO的电子组态
表中的Au5d,6s, 6p和AunCr-1中的Au5d, 6s, 6p, Cr3d, 4s, 4p轨道的有效电荷分布

4 总 结