水热法制备分散型Co促进的MoS2悬浮床加氢脱硫催化剂
2021-12-22王小平马怀军王冬娥田志坚
王小平,马怀军,王冬娥,田志坚
(1.中国科学院 大连化学物理研究所,辽宁 大连 116023;2.中国科学院大学 化学工程学院,北京 100049)
原油资源重质化和劣质化加剧,以及液体燃料品质标准提高,使得石油资源的清洁生产和高效利用面临重大挑战。悬浮床加氢技术是实现劣质重油高效转化的有效技术手段[1-2],该技术已成为重质油加氢领域的研究热点之一。重油的性质和悬浮床加氢工艺要求催化剂同时具有良好的分散和加氢裂化性能,因此高性能催化剂的开发是悬浮床重油加工技术工业应用的技术瓶颈之一。MoS2是目前广泛使用的钼基悬浮床加氢催化剂的活性组分[3-4],但MoS2的活性仍有待提升。
MoS2是一种典型的层状过渡金属硫化物,层间是范德华力相互作用,层内是共价键相互作用。在S-Mo-S分子层中,1个Mo原子与6个S原子成键,而在边缘位1个Mo原子与4个S原子成键,形成S空位。大量研究表明,MoS2的边缘位是催化加氢的活性中心[5-6]。Chianelli等[6]研究了噻吩加氢脱硫中产物选择性与MoS2颗粒层结构的关系,提出了“Rim-edge”模型。在该模型中,将顶层和底层的边缘活性位定义为Rim位,同时具有加氢和脱硫活性;除去顶层和底层的边缘活性位定义为Edge位,只具有脱硫活性;因此通过调控MoS2的堆积层数就可以调变催化剂的加氢脱硫选择性。少层数、纳米级的MoS2催化剂可暴露更多的活性位,有利于催化活性的提高。
大量研究表明:添加Co助剂能显著提高负载型MoS2催化剂的加氢性能[7-8]。Co在MoS2中的助剂效应对Co促进MoS2(Co-Mo-S)催化剂活性的影响,是Co-Mo-S催化剂研发和制备中重要的科学问题。在Co-Mo-S催化剂的制备过程中,Co/Mo摩尔比是重要的影响因素。研究Co/Mo摩尔比对Co-Mo-S催化剂活性影响有助于理解Co-Mo-S催化剂中钴的助剂效应,对进一步提高Co-Mo-S催化剂的活性具有重要的意义。Topsøe等[9-10]在氧化铝负载的Co促进MoS2催化剂中观测到一种活性相结构,并将其命名为“Co-Mo-S”相,该结构模型很好地解释了Co在MoS2催化剂中的助剂效应。Lauritsen等[11]通过扫描隧道显微镜观察到“Co-Mo-S”相结构与MoS2的结构相似,Co原子完全取代MoS2的(1010)面S边相邻内侧的Mo原子,同时“Co-Mo-S”相形貌由MoS2原子簇的三角形变为六边形。与Co原子相邻的S原子的电子环境的改变可能是“Co-Mo-S”相活性增加的关键,且加氢活性依赖于“Co-Mo-S”相的数量。
Co-Mo-S催化剂的活性取决于其纳米结构,而其纳米结构可以通过合成方法调节。合成分散型Co-Mo-S纳米颗粒的常用方法是先通过溶液反应、共沉淀或浸渍等方法制备硫代钼酸盐前体或氧化物前体,然后在H2或H2/H2S气氛中还原硫化制得分散型Co-Mo-S催化剂。Li等[12]用Co的层状羟基盐为前体,用离子交换方法在层间引入钼盐阴离子制备Co-Mo氧化物,在体积分数10%的H2S/H2气氛中于400 ℃还原硫化,制得Co-Mo催化剂。制得的非负载Co-Mo催化剂用于FCC汽油加氢脱硫时,表现出很好的脱硫选择性。Genuit等[13]将乙酸钴和硫代钼酸铵进行溶液反应,形成CoMoS4沉淀,所得CoMoS4在体积分数15%的H2S/H2气氛中于400 ℃硫化4 h,即得Co-Mo-S催化剂。然而在这些合成方法中,最后都需要进行高温硫化还原。
水热/溶剂热法操作简单,条件温和,并且不需要高温硫化还原,因而受到研究者的关注。Yoosuk等[14]将硝酸钴和四硫代钼酸铵混合后,在3.8 MPa氢气气氛下于350 ℃反应2 h,制得了Co促进的MoS2催化剂。他们将MoS2和Co促进的MoS2用于二苯并噻吩(DBT)加氢脱硫反应。研究发现,相同条件下,Co促进的MoS2催化剂催化DBT加氢的反应速率是MoS2催化剂的8倍。Sorribes等[8]将七钼酸铵、硫和乙酸钴加入到去离子水中,随后加入水合肼作为还原剂,水热反应后制得非负载的Co促进的MoS2催化剂。该催化剂用于催化3-硝基苯乙烯选择性加氢反应,3-硝基苯乙烯的转化率大于99%,3-氨基苯乙烯的选择性可达91%,而MoS2催化3-硝基苯乙烯的转化率仅为20%。笔者所在课题组前期工作采用水热/溶剂热法合成了MoS2纳米花[15]、MoS2纳米片[4]、准单层MoS2催化剂[16],将其用于多环芳烃化合物(如蒽、菲)的催化加氢反应,表现出了优异的催化活性。可见,水热/溶剂热法可精确调控合成的MoS2催化剂的结构,以提高活性位的暴露量,获得优异的催化活性。
笔者在所在课题组前期研究工作的基础上,以三氧化钼、硝酸钴和L-半胱氨酸为原料。调变投料的Co/Mo摩尔比,一步水热制备高活性的分散型Co-Mo-S催化剂,通过XRD、ICP-OES、BET、Raman、HRTEM、XPS等表征手段确定催化剂的结构、形貌特征和活性组分。在高压釜式反应器中,以DBT为模型化合物考察Co-Mo-S催化剂的加氢脱硫活性。以通过水热法一步制备高活性和高选择性的Co-Mo-S催化剂为研究目的,研究Co-Mo-S催化剂制备过程中原料中Co/Mo摩尔比对Co-Mo-S催化剂中形貌(层数、片层长度)以及活性相的影响,对Co-Mo-S催化剂的构效关系提出一些新的认识。
1 实验部分
1.1 原料和试剂
三氧化钼(MoO3,质量分数99.5%)、L-半胱氨酸(C3H7NO2S,质量分数99.5%)、二苯并噻吩(C12H8S,质量分数99%)、十氢萘(C10H10,质量分数98%),阿拉丁试剂(上海)有限公司产品;硝酸钴(Co(NO3)2·6H2O,质量分数99%),天津科密欧化学试剂有限公司产品;乙醇(C2H5OH,质量分数99.5%),天津大茂化学试剂厂产品。
1.2 催化剂的制备
水热法制备不同Co/Mo摩尔比的Co-Mo-S催化剂的步骤如下:按照Co/Mo摩尔比分别为0.03、0.10、0.30、1.00、3.00及S/(Mo+Co)原子摩尔比为4的化学计量比称取0.288 g三氧化钼和相应质量的硝酸钴和L-半胱氨酸,加入到50 g去离子水中。以300 r/min速率搅拌30 min后,将该混合物转移至100 mL带有聚四氟乙烯内衬的不锈钢反应釜中,于200 ℃保温24 h。L-半胱氨酸作为硫化剂和还原剂,无需额外添加还原剂,即可实现MoS2的制备。
L-半胱氨酸还可以通过巯基、氨基和羧基基团与Mo6+形成三齿螯合物,使生成的MoS2的尺寸得到有效调控。L-半胱氨酸与三氧化钼的反应方程式如式(1)、式(2)所示。

(1)
(2)
水热反应所得黑色产物以4500 r/min速率离心分离后,用无水乙醇和水各洗涤3次,70 ℃下真空干燥12 h,得到的样品记为Co-Mo-S-x(x代表催化剂中Co/Mo摩尔比)。洗涤后真空干燥的操作不会导致生成的硫化物催化剂被氧化。
另外,水热合成MoS2和CoS2过程同Co-Mo-S催化剂,但不添加钴源,按S/Mo摩尔比为4制备的分散型MoS2催化剂标记为MoS2-H;水热过程不添加钼源,按S/Co摩尔比为4制备的分散型CoS2催化剂记为CoS2-H。
1.3 催化剂的表征
X射线衍射表征采用PANalytical公司Philips X’Pert PRO型X射线衍射仪,Cu靶,Kα射线(λ=0.15418 nm),管电压40 kV,管电流40 mA,扫描范围为5°~70°,扫描速率为13 °/min。用Debye-Scherrer公式Lc=kλ/βcosθ(其中k=0.9,λ=0.15418 nm,β为(002)峰的半高宽(rad),θ为布拉格角(°))来计算(002)峰的平均堆积高度Lc(nm)。用布拉格方程2dsinθ=Nλ(d为(002)峰的晶面间距(nm),N为衍射级数,取N=1,λ=0.15418 nm)来计算(002)峰的层间距d002(nm)。采用美国Micromeritics公司的ASAP 2420物理吸附仪测定催化剂的比表面积。测试前,催化剂在300 ℃下抽真空处理4 h,然后在液氮温度下(-196 ℃)进行氮气吸附-脱附等温线的测定。催化剂的比表面积采用BET方程计算,孔体积采用BJH方法计算。高分辨透射电子显微镜(HRTEM)表征采用JEOL公司JEM-2100 型透射电子显微镜,加速电压为200 kV。拉曼光谱(Raman)使用Renishaw和JPK公司的Nano Wizard Ultra Speed &inVia Raman拉曼-原子力显微镜连用成像分析系统采集,激光波长为532 nm,激光功率为0.5 mW,采集时间为240 s。电感耦合等离子体发射光谱(ICP-OES)采用PerkinElmer公司的Optima 7300DV ICP-OES分析仪进行Co、Mo元素的定量分析,催化剂经浓硝酸溶解并稀释。X射线光电子能谱(XPS)表征采用Thermo ESCALAB 250Xi spectrometer,AlKα单色光源(Ehυ=1486.6 eV)。催化剂全谱采用248.6 eV的C 1 s进行校正,分峰拟合使用XPS PEAK 4.1软件处理。Mo 3d5/2中Mo4+物种的摩尔分数为Mo4+物种的峰面积占Mo4+、Mo5+和Mo6+峰面积之和的百分比,Mo5+和Mo6物种的摩尔分数的计算方式与Mo4+物种相同。
1.4 催化剂的活性评价
称取3.0 g的反应物DBT、0.075 g催化剂、30 g十氢萘溶剂加入100 mL的Parr釜式反应器中。密闭釜体,用0.5~1.0 MPa的氮气置换釜中空气3次,再用0.5~1.0 MPa的氢气置换氮气3次,随后充氢气至压力为8 MPa。调节搅拌转速为300 r/min,以10 °/min的速率升温至350 ℃、自生压力下反应4 h。反应结束后自然冷却,取出反应物进行分析。用孔径为0.22 μm的微孔滤膜过滤掉产物中的催化剂,采用装有HP-5型色谱柱的Agilent公司7890B-5977A型GC-MS气-质联用装置定性分析其成分,采用装有HP-5型色谱柱和FID检测器的Agilent公司7890A型气相色谱仪进行定量分析,通过自测的相对质量校正因子进行色谱定量计算。
二苯并噻吩转化率x(%)使用公式(3)计算。
x=(m0-m1)/m0×100%
(3)
式中:m0、m1分别为二苯并噻吩的初始质量和反应后剩余的质量,g。
二苯并噻吩加氢脱硫(HDS)的典型反应路径,一是直接脱硫生成主产物联苯(BP)的直接脱硫反应路径(DDS);二是先加氢生成四氢化二苯并噻吩(THDBT),再加氢脱硫生成苯基环己烷(CHB)的加氢路径(HYD)[17]。此外,BP和CHB还可进一步发生加氢裂化或加氢异构反应生成甲基环戊烷(MCP)、苯(PhH)。液相某一产物的摩尔选择性si(%)和收率yi(%)分别用公式(4)和(5)计算。
si=ni/∑ni×100%
(4)
yi=x×si
(5)
式中:ni为i组分的物质的量,mol;x为二苯并噻吩转化率,%。
DDS路径与HYD路径选择性之比用公式(6)计算。
sDDS/sHYD=sBP/(sTHDBT+sCHB+sBCH)
(6)
式中:sDDS/sHYD为DDS路径与HYD路径选择性之比,sBP、sTHDBT、sCHB、sBCH分别为BP、THDBT、CHB、BCH的选择性。
2 结果与讨论
2.1 催化剂的表征
2.1.1 催化剂的XRD表征

2H-MoS2的(002)峰衍射峰强度与其在c轴方向的堆积层数有关。由Debye-Scherrer公式计算出MoS2-H、Co-Mo-S-0.03、Co-Mo-S-0.1、Co-Mo-S-0.3和Co-Mo-S-1的(002)峰的平均堆积高度在3.9~4.2 nm,除以片层的层间距(d002=0.96 nm),可以计算出其片层平均堆积层数分别为4.2、4.3、4.4、4.1和3.9。由此可知,与MoS2-H相比,Co的加入没有改变Co/Mo摩尔比不大于1的Co-Mo-S催化剂中片层的堆积层数。

(1)MoS2-H;(2)Co-Mo-S-0.03;(3)Co-Mo-S-0.1;(4)Co-Mo-S-0.3;(5)Co-Mo-S-1;(6)Co-Mo-S-3;(7)CoS2-H图1 MoS2-H、CoS2-H和不同Co/Mo摩尔比的Co-Mo-S催化剂的XRD谱图Fig.1 XRD patterns of MoS2-H,CoS2-H and the Co-Mo-S catalyst with different Co/Mo molar ratios
2.1.2 催化剂的N2等温吸附-脱附和ICP-OES表征
用氮气等温吸附-脱附法表征了MoS2-H、CoS2-H和不同Co/Mo摩尔比的Co-Mo-S催化剂的物理性质,如图2所示。由图2可知:根据IUPAC分类,7种催化剂的N2吸附-脱附等温线均属于Ⅳ型,且具有H3或H4型回滞环。7种催化剂的吸附等温线在p/p0<0.01时无吸附,在p/p0<0.8时缓慢上升,而到p/p0>0.9后吸附快速上升,说明催化剂均没有微孔,只有相互堆积产生的堆积孔[23]。表1 总结了MoS2-H、CoS2-H和不同Co/Mo摩尔比的Co-Mo-S催化剂的组成和结构特征。由表1可知,催化剂的比表面积均小于20 m2/g,孔体积均小于0.1 cm3/g,这可能是由于催化剂的粒径较小且轻微团聚造成的。表1中的ICP-OES数据表明,水热制备的Co-Mo-S催化剂的Co/Mo摩尔比与投料Co/Mo摩尔比一致,说明Co-Mo-S催化剂的Co/Mo摩尔比可以通过投料调控。

(1)MoS2-H;(2)Co-Mo-S-0.03;(3)Co-Mo-S-0.1;(4)Co-Mo-S-0.3;(5)Co-Mo-S-1;(6)Co-Mo-S-3;(7)CoS2-H图2 MoS2-H、CoS2-H和不同Co/Mo摩尔比的Co-Mo-S催化剂的N2吸附-脱附等温线Fig.2 N2adsorption-desorption isotherms of MoS2-H,CoS2-H and the Co-Mo-S catalyst with different Co/Mo molar ratios

表1 MoS2-H、CoS2-H和不同Co/Mo摩尔比的Co-Mo-S催化剂的组成和结构特征Table 1 Composition and structural characteristics of MoS2-H,CoS2-H and the Co-Mo-S catalyst with different Co/Mo molar ratios
2.1.3 催化剂的HRTEM表征
图3为MoS2-H、CoS2-H和不同Co/Mo摩尔比的Co-Mo-S催化剂的HRTEM照片。由图3可知,MoS2-H和Co/Mo摩尔比不大于1的Co-Mo-S催化剂的HRTEM照片中呈现出很多层间距为0.94 nm 的纳米片层,这是MoS2的典型层状结构,对应于层间距扩大的2H-MoS2(002)晶面;Co-Mo-S-3和CoS2-H中间距为0.32 nm的条纹对应于CoS2的(111)晶面。MoS2-H催化剂的HETEM照片中,MoS2由纳米片层堆积而成,统计其堆积层数为3~6层,S-Mo-S 分子层层间距为0.94 nm,片层长度大于40 nm。Co/Mo摩尔比不大于1的Co-Mo-S催化剂具有典型的2H-MoS2片层结构,其堆积层数为3~6层,分子层层间距为0.94 nm,片层长度5~20 nm。随着Co-Mo-S催化剂中Co/Mo摩尔比增加,Co-Mo-S 催化剂片层长度变化不显著,这是因为,部分Co单独成相,没有掺杂进MoS2的边缘。相比于MoS2-H催化剂,Co-Mo-S催化剂中片层的长度变短,但堆积层数和层间距基本不变,与XRD结果一致。这是由于部分Co取代MoS2边缘的Mo后,形成“Co-Mo-S”相,导致其边缘出现缺陷,阻碍了MoS2基底方向的生长,从而使片层长度变短。Berhault等[24]认为,Co最大限度地与MoS2边缘相互作用,可以作为硫化钼相邻两层的“黏合剂”,从而使MoS2的堆叠层数保持稳定或增加。所以Co的引入没有改变MoS2片层的堆积层数,但使其片层长度变短。在Co-Mo-S-0.03、Co-Mo-S-0.1 中未观察到明显的CoS2的条纹,说明Co均匀地掺杂在MoS2中。在Co-Mo-S-0.3、Co-Mo-S-1中也未观察到明显的CoS2的条纹,但在其对应样品的XRD谱图中可以看到归属于CoS2的特征衍射峰,说明部分Co均匀地掺杂在MoS2中。在Co-Mo-S-3催化剂中可以观察到CoS2晶体的边缘位置有片层数为1~2层的MoS2附着(图3(f)中白色圆圈所示),此时CoS2可视为MoS2的载体。

图3 MoS2-H、CoS2-H和不同Co/Mo摩尔比的Co-Mo-S催化剂的HRTEM图像Fig.3 HRTEM images of MoS2-H,CoS2-H and the Co-Mo-S catalyst with different Co/Mo molar ratios(a)MoS2-H;(b)Co-Mo-S-0.03;(c)Co-Mo-S-0.1;(d)Co-Mo-S-0.3;(e)Co-Mo-S-1;(f)Co-Mo-S-3;(g)CoS2-H
2.1.4 催化剂的Raman表征


(1)MoS2-H;(2)Co-Mo-S-0.03;(3)Co-Mo-S-0.1;(4)Co-Mo-S-0.3;(5)Co-Mo-S-1;(6)Co-Mo-S-3;(7)CoS2-H图4 MoS2-H、CoS2-H和不同Co/Mo摩尔比的Co-Mo-S催化剂的Raman谱图Fig.4 Raman spectra of MoS2-H,CoS2-H and the Co-Mo-S catalyst with different Co/Mo molar ratios

2.1.5 催化剂的XPS表征

Pimerzin等[30]研究了CoMo/Al2O3的活性与“Co-Mo-S”相的质量分数之间的关系,他们将“Co-Mo-S”相的相对含量与XPS测的Co的质量分数相乘,得到催化剂表面的“Co-Mo-S”相的质量分数,CoMo/Al2O3催化噻吩加氢脱硫的反应速率与“Co-Mo-S”相的质量分数表现出非常好的相关性。参考文献[30]的计算方法,得到催化剂中“Co-Mo-S”相的质量分数,如表2所示。由表2可以看出,随着Co-Mo-S催化剂中Co/Mo摩尔比增加,“Co-Mo-S”相的质量分数先增加后减小。Co-Mo-S-1催化剂中的“Co-Mo-S”相质量分数最高,预示着Co-Mo-S-1催化剂会有最好的活性。
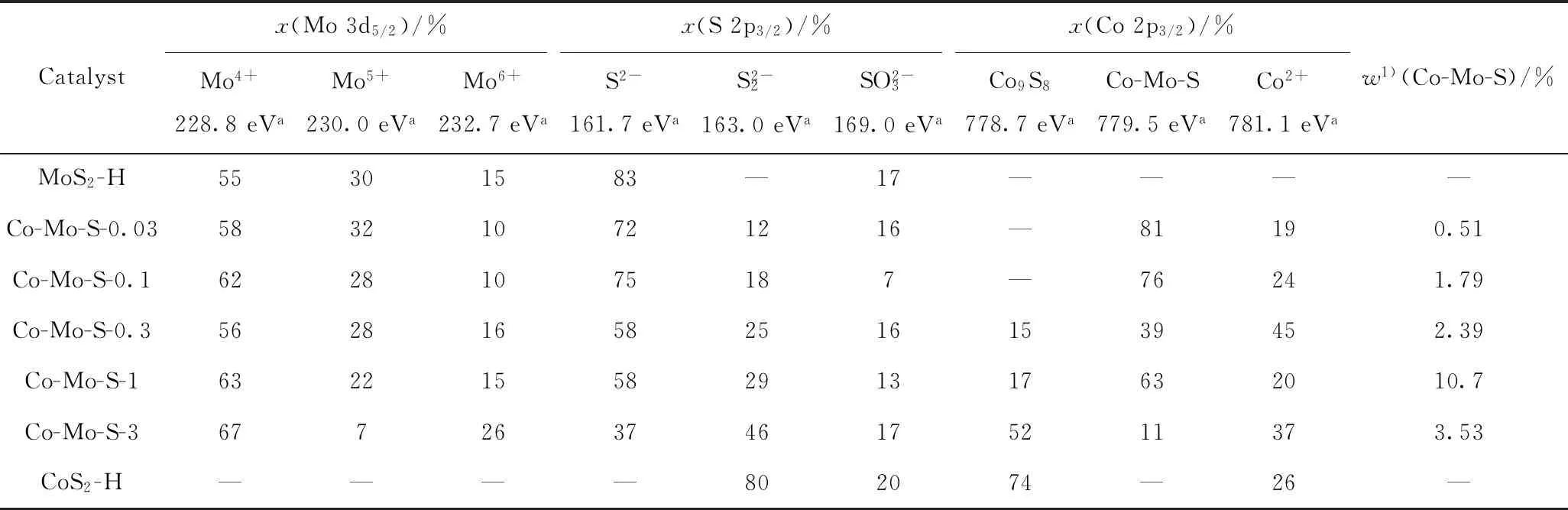
表2 MoS2-H、CoS2-H和不同Co/Mo摩尔比的Co-Mo-S催化剂的XPS定量分析结果Table 2 XPS quantitative analysis results of MoS2-H,CoS2-H and the Co-Mo-S catalyst with different Co/Mo molar ratios
综合以上表征可知,Co/Mo摩尔比不大于0.1的Co-Mo-S催化剂中,部分Co取代MoS2的S-Mo-S层中的钼形成“Co-Mo-S”相,随着Co/Mo摩尔比增加,部分Co形成“Co-Mo-S”相,另一部分Co形成CoS2晶体,CoS2和MoS2两相共存。当Co/Mo摩尔比增加至3时,Co-Mo-S催化剂中主要为表面有MoS2附着的CoS2。Co/Mo摩尔比不大于1的Co-Mo-S催化剂中,Co助剂的加入一方面形成了高活性的“Co-Mo-S”相,另一方面Co助剂虽没有改变片层的堆积层数,但造成片层长度变短,增加了边位的数量,暴露出更多活性位,从而可能使得Co-Mo-S 催化剂具有高加氢活性。
2.2 催化剂的DBT加氢评价
硫是重油中主要的杂原子和杂质之一,以各种有机硫化物(如噻吩,二苯并噻吩)的形式存在于重质油中,噻吩硫占重油中总硫的80%以上[31]。二苯并噻吩(DBT)是一种较难脱硫的含硫稠环化合物,而含硫稠环化合物是重油的主要特征分子[32]。含硫稠环化合物的脱硫可以促进大分子键的打开,进而发生裂解,实现重油轻质化。因此采用二苯并噻吩作为重质油模型化合物,以高压釜式反应器模拟悬浮床,对MoS2-H、CoS2-H和不同Co/Mo摩尔比的Co-Mo-S催化剂的加氢脱硫性能进行评价,结果如图5所示。
DBT加氢脱硫(HDS)典型反应路径为DDS路径和HYD路径,DDS路径每脱除1个S原子需消耗2个氢气分子,而HYD路径则需消耗4个氢气分子,因此提高DDS路径的选择性有利于减少反应的氢耗。由图5(a)可知:随着Co-Mo-S催化剂中Co/Mo摩尔比增加,DBT的转化率和sDDS/sHYD均先增加后减小,呈现典型的火山形曲线。但Co-Mo-S催化剂加氢脱硫活性与选择性并不完全一致,如Co-Mo-S-3催化剂上DBT的转化率低于Co-Mo-S-0.3催化剂上DBT的转化率,但Co-Mo-S-3催化剂上的sDDS/sHYD却高于Co-Mo-S-0.3催化剂上的sDDS/sHYD。Co-Mo-S催化剂催化DBT加氢的转化率和sDDS/sHYD均高于MoS2-H催化剂,其中Co-Mo-S-1催化剂上的DBT转化率和sDDS/sHYD分别为88.4%和0.9,分别是MoS2-H催化剂上的1.8倍和9倍。以上DBT加氢评价结果说明,Co-Mo-S催化剂具有优异的催化DBT加氢的性能,并且随着Co-Mo-S催化剂中Co/Mo摩尔比的增加催化剂的活性呈现火山形曲线。
图5(b)是MoS2-H、CoS2-H和不同Co/Mo摩尔比的Co-Mo-S催化剂上DBT加氢产物的收率。随着Co-Mo-S催化剂中Co/Mo摩尔比增加,直接脱硫产物BP和裂解产物PhH与MCP的收率均先增加后减小;而加氢路径产物THDBT的收率先减小后增加,CHB的收率先增加后减小,BCH的收率一直减小。Co-Mo-S-1催化剂BP和PhH的收率为34.9%和10.9%,分别是MoS2-H上的7.3倍和5.9倍。以上结果说明,与MoS2-H相比,Co-Mo-S 催化剂具有优异的直接脱硫性能和加氢裂解性能。
随着Co-Mo-S催化剂中Co/Mo摩尔比的增加,“Co-Mo-S”相的质量分数先增加后减小,这与DBT转化率、sDDS/sHYD以及BP收率的变化规律一致。说明Co/Mo摩尔比不大于1的Co-Mo-S催化剂的活性与“Co-Mo-S”相的含量呈现正相关,“Co-Mo-S”相是主要的活性位。而Co-Mo-S-3催化剂中“Co-Mo-S”相的质量分数高于Co-Mo-S-0.3催化剂中“Co-Mo-S”相的质量分数,Co-Mo-S-3催化剂的活性却低于Co-Mo-S-0.3催化剂,这是由于反应评价时采用的催化剂质量都是0.075 g,相同质量的Co-Mo-S-0.3催化剂和Co-Mo-S-3催化剂中,前者含有的MoS2多,后者含有的CoS2多,而MoS2的加氢活性高于CoS2。

DDS—Direct desulfurization;HYD—Hydrogenation;DBT—Dibenzothiophene;THDBT—Tetrahydrodibenzothiophene;BP—Biphenyl;CHB—Phenylcyclohexane;BCH—Bicyclohexyl;MCP—Methylcyclopentane;PhH—Benzene图5 MoS2-H、CoS2-H和不同Co/Mo摩尔比Co-Mo-S催化剂上的加氢脱硫性能评价Fig.5 Hydrodesulfurization performance evaluation of MoS2-H,CoS2-H and the Co-Mo-S catalyst with different Co/Mo molar ratios(a)Conversion of DBT and selectivity ratio of DDS/HYD on MoS2-H,CoS2-H and the Co-Mo-S catalysts with different Co/Mo molar ratios;(b)Yields of DBT hydrodesulfurization products on MoS2-H,CoS2-H and the Co-Mo-S catalysts with different Co/Mo molar ratios Reaction conditions:mDBT=3 g;mcatalyst=0.075 g;msolvent-decalin=30 g;T=350 ℃;p(initial H2)=8 MPa;t=4 h
3 结 论
以三氧化钼、硝酸钴和L-半胱氨酸为原料,一步水热法制得了片层短、层数少、含有高活性“Co-Mo-S”相的Co-Mo-S催化剂。
(1)当Co/Mo摩尔比不大于1时,Co-Mo-S催化剂均为纳米片层堆积而成,部分Co取代MoS2的S-Mo-S层中的Mo形成“Co-Mo-S”相,片层层间距为0.94 nm,平均堆积层数约4层,片层长度为5~20 nm。Co/Mo摩尔比进一步增加到3时,Co-Mo-S 催化剂主要为表面层数为1~2层MoS2附着的CoS2晶体。
(2)Co助剂的引入一方面在Co-Mo-S催化剂中形成了高活性的“Co-Mo-S”相;另一方面Co助剂虽没有改变Co-Mo-S催化剂片层的堆积层数,但造成片层长度变短,增加了边位的数量,暴露出更多活性位,从而使得Co-Mo-S催化剂具有高加氢活性。
(3)当Co/Mo摩尔比不大于1时,随着Co-Mo-S催化剂中Co/Mo摩尔比增加,DBT转化率、sDDS/sHYD以及BP收率也增加,这与Co-Mo-S催化剂中“Co-Mo-S”相的质量分数变化规律一致。说明Co-Mo-S催化剂的活性和DDS路径的选择性与“Co-Mo-S”相的含量呈现正相关的关系,“Co-Mo-S”相是Co-Mo-S催化剂主要的活性位。因此,提高Co-Mo-S催化剂的活性,需要提高Co-Mo-S催化剂中“Co-Mo-S”相的含量和减小催化剂的尺寸(层数、片长)。
