依折麦布固体脂质纳米粒的制备与大鼠体内药动学研究
2021-09-10邱爱丽祁静波
邱爱丽,王 敏,谢 鹏,祁静波
(1.唐山市协和医院药剂科,唐山 063000;2.唐山职业技术学院,唐山 063000)
依折麦布通过选择性抑制肠道内胆固醇转运蛋白活性以达到降低胆固醇吸收的目的,是目前唯一的胆固醇吸收抑制剂[1-2]。依折麦布属于生物药剂学分类系统(BCS)Ⅱ类药物,在水中极难溶解(约为2.3 μg·mL-1),不利于药物口服吸收[3-4],另外依折麦布是P-糖蛋白(P-gp)的底物,口服吸收后在P-gp作用下外排,降低了口服生物利用度,增加了药物生物变异性[5],严重影响其治疗效果。本研究将依折麦布制备成固体脂质纳米粒,同时加入D-α-维生素E聚乙二醇1000琥珀酸酯(TPGS)对依折麦布固体脂质纳米粒(EZE-SLNs)表面进行修饰,通过大鼠体内药动学评估药物生物利用度,为依折麦布的口服新剂型研究提供依据。
1 仪器与材料
1.1仪器 安捷伦1220型高效液相色谱仪(安捷伦科技有限公司);DF-101SZ型数显转速集热式恒温加热磁力搅拌器(上海凌科实业发展有限公司);Scientz-1200E型超声波细胞粉碎机(宁波新芝生物科技股份有限公司);JEM-1400Flash型透射电子显微镜(日本电子株式会社);Malvern Zetasizer Nano-ZS型纳米粒径/电位分析仪(英国马尔文公司)。
1.2试药 依折麦布原料药(无锡福祈制药有限公司,批号20191214,质量分数为99.5%);伊曲康唑(中国食品药品检定研究院,批号100631-201402);山嵛酸甘油酯(Compritol-888 ATO,德国巴斯夫公司);大豆磷脂(南京威尔药业股份有限公司);D-α-维生素E聚乙二醇1000琥珀酸酯(TPGS,江西联陆生物科技有限公司);盐酸和无水乙醇,均购自南京化学试剂股份有限公司;磷钨酸钠(上海阿拉丁生化科技股份有限公司);聚乙烯吡咯烷酮K30(PVP K30)和十二烷基硫酸钠,均购自巴斯夫新材料有限公司;pH6.8磷酸盐缓冲液(PBS,自制)。
1.3动物 Wister种大白鼠,雌雄各半,体质量为(200±20) g,天津医科大学提供,合格证号:SYXK(津)2019-004。
2 方法与结果
2.1EZE-SLNs与基于TPGS表面修饰的EZE-SLNs(TPGS-EZE-SLNs)的制备 通过前期的制备工艺筛选,采用乳化超声-低温固化法[6-7]制备EZE-SLNs。按照处方量称取山嵛酸甘油酯2 500 mg加入到乙醇10 mL中搅拌溶解,再称取依折麦布原料药250 mg加入到上述乙醇中搅拌溶解,呈透明状液体,得到油相溶液;另称取大豆磷脂1 000 mg加入到纯化水50 mL中,加热至75 ℃,搅拌分散形成乳白色液体,得到水相溶液;在3 000 r·min-1磁力搅拌下,将上述油相溶液通过注射器滴加到水相溶液中,形成乳白色液体,减压蒸发除去乙醇,加水定容至50 mL;将超声探头置于该溶液中进行超声处理,超声功率为200 W,超声10 s,间歇5 s,持续超声5 min,超声结束后立即将溶液置于冰水浴中冷却,即得到EZE-SLNs。除在水相中加入TPGS 200 mg外,TPGS-EZE-SLNs的制备工艺和处方中各成分的用量与EZE-SLNs均相同。将制备得到的EZE-SLNs与TPGS-EZE-SLNs置于4 ℃冰箱中冷藏,备用。
2.2制剂性质表征
2.2.1粒径分布/Zeta电位测定 使用Malvern Zetasizer Nano-ZS型纳米粒径/电位分析仪测量EZE-SLNs与TPGS-EZE-SLNs的粒径分布、多聚分散系数(PDI)和Zeta电位。样品的制备方法:取EZE-SLNs与TPGS-EZE-SLNs各200 μL,分别置于直径为1 cm的石英比色杯中,加入去离子水3 mL稀释,轻轻振摇,置于激光粒度测定仪中检测,检测波长为635 nm,入射角为90°,环境温度为25 ℃,每个样品平行测定3次,取平均值,结果见表1。
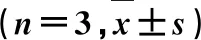
表1 制剂性质检测结果
EZE-SLNs与TPGS-EZE-SLNs的粒径大小相近,均在100 nm左右,文献报道[8-9],纳米粒子在100 nm左右更容易透过肠道细胞进入体内;Zeta电位是评价胶体分散体稳定性的重要指标,其绝对值越大表明该体系越稳定,固体脂质纳米粒能够维持稳定性需要Zeta电位绝对值大于30 mV,只有达到该电位绝对值,扩散层才具有足够的厚度避免微粒之间聚集[10]。本研究制备的2种固体脂质纳米粒Zeta电位绝对值均大于30 mV,且TPGS-EZE-SLNs的Zeta电位绝对值更大,测定的电位值能够间接表明EZE-SLNs与TPGS-EZE-SLNs物理稳定性良好,不易发生聚集和沉淀。
2.2.2微观形态观察 通过透射电子显微镜观察EZE-SLNs与TPGS-EZE-SLNs的表面形态。分别取EZE-SLNs和TPGS-EZE-SLNs各1滴,滴加到碳涂层的铜网上,再滴加蒸馏水稀释,挥干水分,滴加质量浓度为10 mg·mL-1的磷钨酸溶液负染色,再次挥干水分,在透射电镜下观察2个样品的微观形态。见图1。

图1 EZE-SLNs(A)和TPGS-EZE-SLNs(B)的透射电镜照片
由图1可知,EZE-SLNs和TPGS-EZE-SLNs均呈球形分布,边界清晰,无药物结晶析出,粒径大部分在50~100 nm之间。
2.3溶出度比较 载药固体脂质纳米粒口服进入胃肠道后通过内吞作用进入体循环,因此,需要模拟体内环境考察固体脂质纳米粒在pH值为1.2和pH值为6.8条件下的体外药物释放情况,以确定纳米粒在被吸收进入体循环之前药物从纳米载体中释放到释放介质中的程度。采用动态透析法比较EZE-SLNs、TPGS-EZE-SLNs和EZE混悬液的药物释放度。选择截留相对分子质量为10 kDa的透析膜,将2种固体脂质纳米粒样品溶液及EZE混悬液(药物分散到PVP K30溶液中)置于透析袋中,两端系紧,药物含量均为10 mg,释放介质在前2 h选择pH值为1.2的盐酸溶液(含十二烷基硫酸钠质量浓度为5 mg·mL-1),2 h后选择pH值为6.8的PBS(含十二烷基硫酸钠质量浓度为5 mg·mL-1),介质体积均为500 mL,水浴温度为37 ℃,搅拌速度为100 r·min-1,在预定的时间间隔(0、0.5、1、2、4、6、8、12 h)取样,经0.45 μm微孔滤膜过滤,取续滤液适当稀释后使用HPLC法进行分析。每个时间点取样后立即将相同体积的空白释放介质[温度保持在(37±0.5) ℃]补加到溶出杯中。释放曲线见图2。
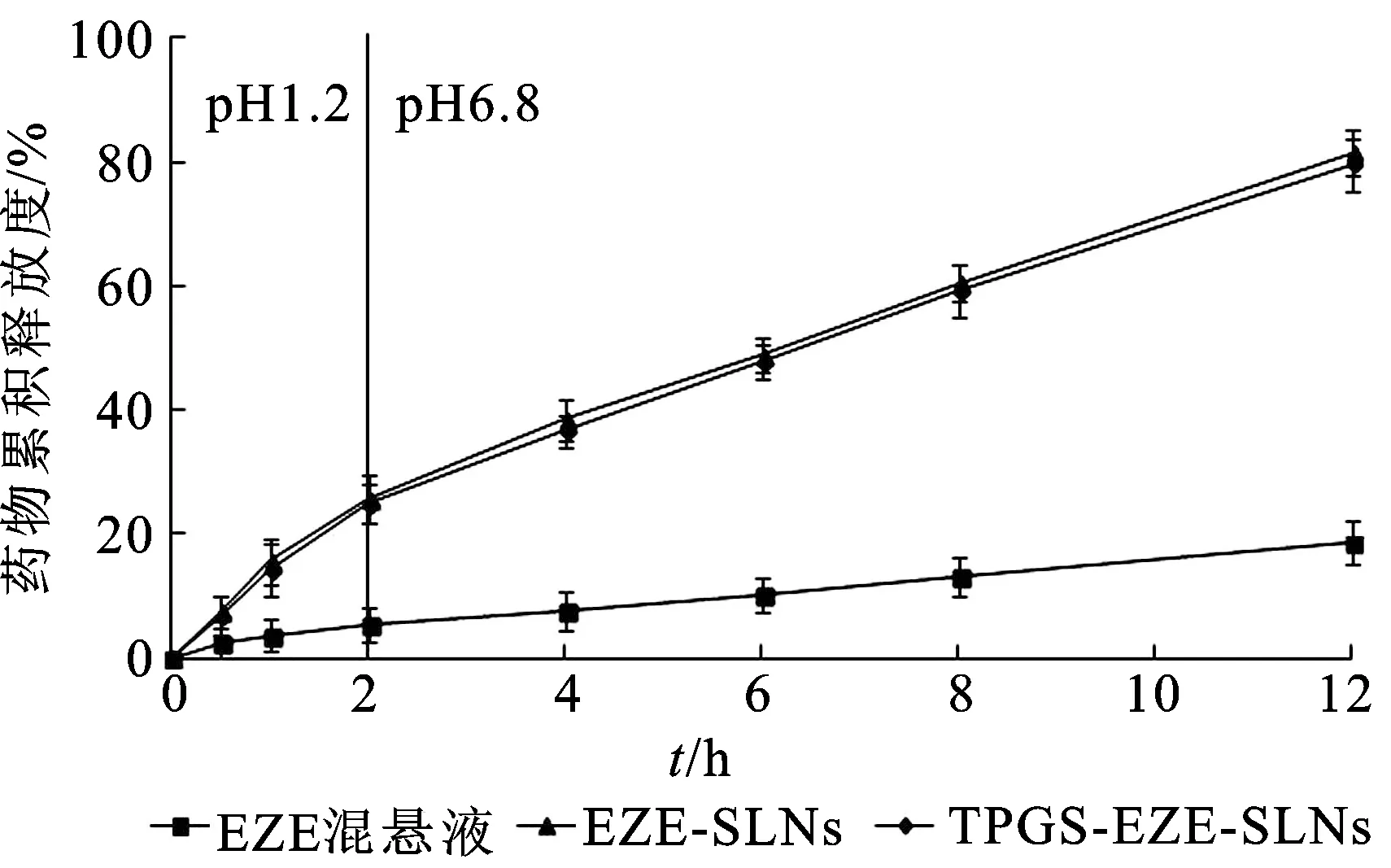
图2 EZE-SLNs、TPGS-EZE-SLNs和EZE混悬液的体外药物释放曲线
由图2可知,EZE-SLNs和TPGS-EZE-SLNs前2 h在pH值为1.2的盐酸中的释放量基本一致,约有25%的药物从载药固体脂质纳米粒中释放出来,随后置于pH值为6.8 PBS中,在12 h约有80%的药物释放;EZE混悬液药物释放较为缓慢,前2 h在pH值为1.2的盐酸中仅有约5%的药物释放,在pH值为6.8的PBS中12 h药物释放量也仅仅达到约18%。实验结果表明,与EZE混悬液相比,EZE-SLNs和TPGS-EZE-SLNs均能提高药物的释放度,有望改善药物的生物利用度。
2.4模拟人体生理环境稳定性考察 固体脂质纳米粒在胃肠道内滞留一段时间才能被吸收进入体内,因此,胃肠道中的液体环境会对纳米粒的稳定性产生一定的影响,进而阻碍药物吸收[11]。因此本研究考察EZE-SLNs和TPGS-EZE-SLNs在模拟人工胃液(SGF)或模拟人工肠液(SIF)中的粒径及Zeta电位变化,结果见图3和图4。
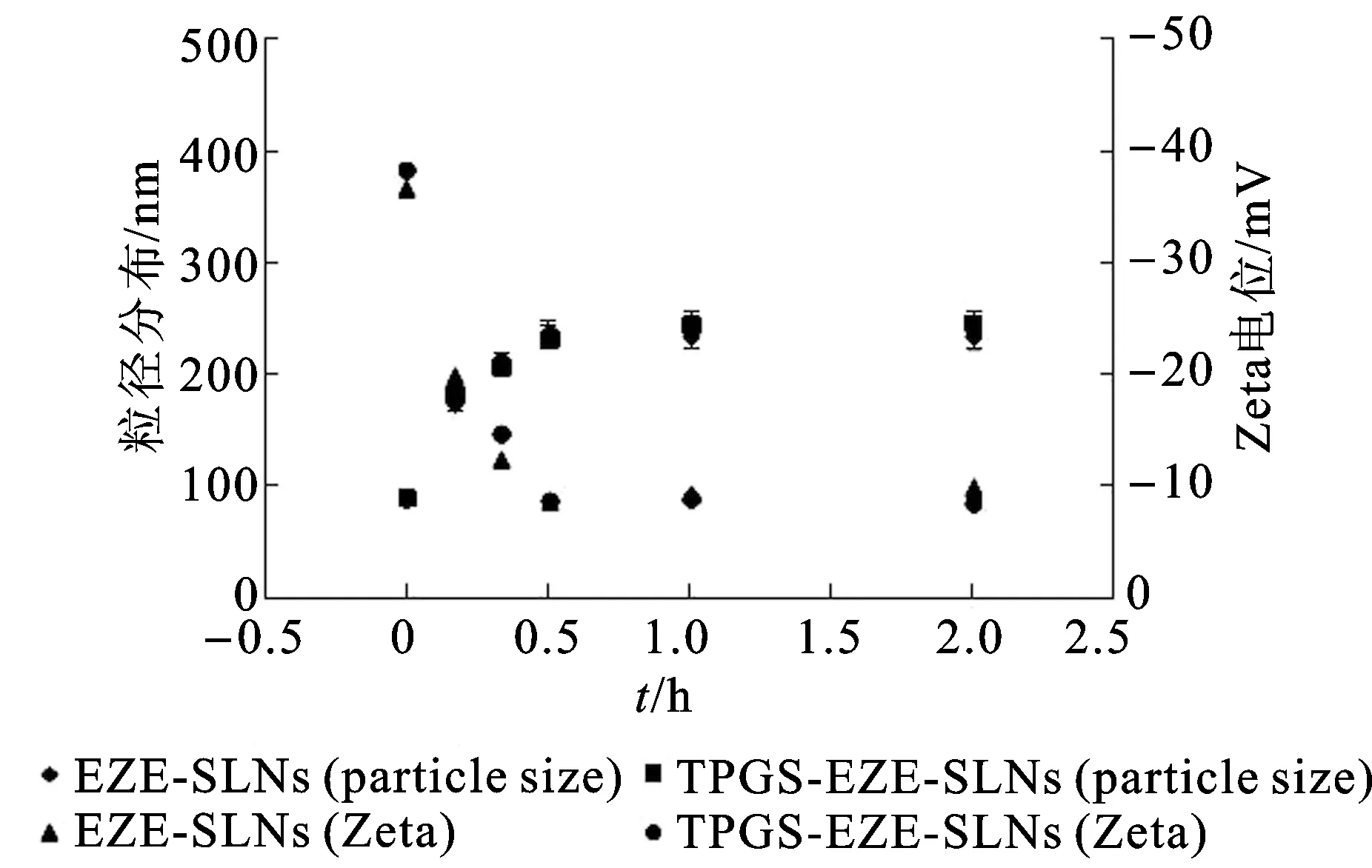
图3 EZE-SLNs和TPGS-EZE-SLNs在SGF中的粒径及Zeta电位变化
图4 EZE-SLNs和TPGS-EZE-SLNs在SIF中的粒径及Zeta电位变化
由图3和图4可知,EZE-SLNs和TPGS-EZE-SLNs在SGF中前30 min粒径呈增加趋势,平均直径均达到200~300 nm,随后粒径不再增加,测得固体脂质纳米粒的Zeta电位为-8~-10 mV,Zeta电位绝对值降低,导致纳米粒之间发生聚集,粒径增大。相反,EZE-SLNs和TPGS-EZE-SLNs在SIF中的粒径基本保持不变,测得Zeta电位显示与初始值相近,为-35~-38 mV。
2.5药动学研究
2.5.1色谱条件 色谱柱为安捷伦Eclipse XDB-C18柱(150 mm×4.6 mm,5 μm);流动相为水-乙腈(30∶70);流速为1.0 mL·min-1;柱温为30 ℃;进样量为20 μL;检测波长为232 nm[12]。
2.5.2血浆处理 大鼠眼眶取血,用内壁涂有肝素钠的圆底离心管收集血液,以4 000 r·min-1离心10 min,收集血浆,取大鼠血浆150 μL,置于尖底离心管中,加入质量浓度为100 mg·L-1的伊曲康唑甲醇溶液10 μL作为内标,涡旋混合5 min,加入乙腈200 μL,再涡旋混合,沉淀血浆蛋白,以10 000 r·min-1离心10 min,取上清液20 μL,按照2.5.1项下色谱条件检测药物含量,计算药物质量浓度。
2.5.3标准曲线 精密称取依折麦布对照品10.0 mg,置于100 mL量瓶中,加入甲醇溶解并稀释定容,摇匀,得依折麦布对照品储备液(含依折麦布为100 mg·L-1)。将上述对照品储备液稀释成依折麦布质量浓度分别为1.0、2.0、4.0、10.0、20.0、40.0 mg·L-1,得系列质量浓度的依折麦布甲醇对照品溶液。取空白大鼠血浆150 μL,置于尖底离心管中,精密加入上述系列质量浓度的依折麦布甲醇对照品溶液10 μL,配制成含有依折麦布质量浓度为0.05、0.1、0.2、0.5、1.0、2.0、5.0 mg·L-1的血浆样品,涡旋混合5 min,再按照2.5.2项下血浆处理方法操作,按照2.5.1项下色谱条件检测药物含量,以峰面积(A)为纵坐标、药物质量浓度(C)为横坐标,采用加权最小二乘法进行回归,得方程:A=63.564C-1.316(r=0.999 2),线性关系良好。
2.5.4精密度及提取回收率 按照2.5.3项下标准曲线方法,取150 μL空白大鼠血浆,置于尖底离心管中,精密加入10 μL系列质量浓度的依折麦布甲醇对照品溶液,配制成含有依折麦布质量浓度分别为0.05、0.5、5.0 mg·L-1的溶液,涡旋混合5 min,再按照2.5.2项下血浆处理方法操作,按照2.5.1项下色谱条件检测药物含量,考察方法精密度和提取回收率。结果显示,上述低、中和高3种药物质量浓度的提取回收率分别为92.1%、95.2%、97.2%,精密度RSD值为3.9%,说明本方法提取回收率较高,精密度较好,符合方法要求。
2.5.5药动学实验 取18只体质量为180~220 g的Wistar大鼠,雌雄各半,随机分为A、B和C 3组,每组6只,实验前将大鼠禁食12 h,自由饮水。A组大鼠灌胃给予EZE混悬液(药物分散到PVP K30溶液中),B组大鼠灌胃给予EZE-SLNs,C组大鼠灌胃给予TPGS-EZE-SLNs,给药剂量均为5 mg·kg-1,在给药后0.25、0.50、0.75、1、2、3、4、6、8、10、12、24 h时间点采用毛细管眼眶取血,每次收集约0.5 mL血样,置于内壁涂有肝素钠的尖底离心管中,以4 000 r·min- 1离心10 min,取上层血浆于-20 ℃冷冻保存。血浆样品按照2.5.2项下血浆处理方法操作,按照2.5.1项下色谱条件检测药物含量,使用DAS 2.0药动学分析软件计算以下药动学参数:达峰时间(tmax)、达峰质量浓度(Cmax)、药时曲线下面积(AUC0~∞)、半衰期(t1/2)和消除速率(CL),并采用t检验分析不同给药组之间的差异性,P<0.05表示差异具有统计学意义。

表2 大鼠药动学参数

图5 平均血药质量浓度-时间曲线
3 讨论
为了增加药物溶解度以及提高生物利用度,研究人员尝试将依折麦布制备成固体分散体[13]、介孔二氧化硅载药[14]和自乳化释药系统[15-16]等新型给药系统。固体脂质纳米粒是20世纪90年代开始研究的较为成熟的一种纳米药物给药系统,是由生物相容性良好的天然或合成脂质和表面活性剂构成,药物包裹在纳米粒的脂质内部或吸附在纳米粒表面,SLNs属于纳米胶态分散体系,粒径在50~1 000 nm之间,能够显著提高药物的溶解度和溶出速率,是极具开发潜力的纳米粒给药系统[17-18]。TPGS属于非离子表面活性剂,文献报道[19-20],TPGS能够抑制P-gp对底物的外排作用,改变生物膜的流动性,提高药物的生物利用度,作为一种安全的药用辅料已被美国食品和药物管理局(FDA)批准用于药品中。
由大鼠体内药动学研究结果可知,与EZE混悬剂组相比,EZE-SLNs组和TPGS-EZE-SLNs组的AUC0~∞显著增加,分别是EZE混悬剂组的1.76倍和2.08倍,说明大鼠口服EZE-SLNs和TPGS-EZE-SLNs可以显著提高药物的生物利用度;EZE-SLNs组和TPGS-EZE-SLNs组的Cmax分别是EZE混悬剂组的1.44倍和1.81倍,2种固体脂质纳米粒均提高了药物的Cmax,这可归因于依折麦布制备成固体脂质纳米粒后,粒径急剧减小,比表面积急剧增大,改善了药物的溶解性,在进入体内后能够快速、充分地被十二指肠及小肠上皮细胞吸收。此外,TPGS-EZE-SLNs组的AUC0~∞与Cmax分别是EZE-SLNs组的1.19倍和1.26倍,说明TPGS-EZE-SLNs提高药物生物利用度更显著,这归因于TPGS-EZE-SLNs处方中的TPGS抑制了肠上皮细胞膜中的P-gp的活性,降低了对依折麦布的外排作用,提高了药物的生物利用度。
