LC-MS/MS 用于头孢拉定胶囊强制降解产物的结构分析
2021-07-19黄道明郑露盼张继红
黄道明,郑露盼,张继红
上饶市食品药品检验检测中心,江西 上饶 334000
头孢拉定是第一代半合成头孢菌素,结构式见图1。其合成方法以化学合成法为主:即以7-氨基去乙酰氧基头孢烷酸(7-ADCA)经成盐或酯化后,与经保护后的双氢苯甘氨酸中间体缩合,再水解、结晶得到头孢拉定[1]。随着对药品杂质特性的深入研究,依据杂质的药理特性,逐一制定每一个杂质限度的“杂质谱控制理念”已经被国内外普遍接受[2-5]。本研究前期采用了HPLC 法测定头孢拉定胶囊强制降解产物,明确了头孢拉定胶囊粉末在酸、碱及氧化条件下均有明显的降解产物生成[6]。而本次研究根据杂质谱控制理念,运用强制降解试验,进一步对头孢拉定胶囊强降解产物的结构及来源进行推测,为头孢拉定制剂稳定性研究提供重要依据,为药品生产、运输及贮存条件提供重要参考[7-11]。

图1 头孢拉定化学结构
1 仪器与试药
1.1 仪器
WatersTQ-S 液相质谱仪(美国沃特世),电子天平(Sartorius 公司),恒温水浴锅(北京市永光明医疗仪器有限公司),pH 计(Thermo 公司)。
1.2 试剂试药
头孢拉定胶囊(A 企业,批号:200310,250 mg/粒);头孢拉定对照品(中国食品药品检定研究院,批号:130427-201107,含量88.3%),头孢氨苄对照品(中国食品药品检定研究院,批号:130408-201411,含量94.4%);乙腈、甲醇为质谱纯(德国EMD Millipore 公司),甲酸(Formic Acid),水(市售屈臣氏饮用水)。
2 方法
2.1 色谱条件
采用SuperLu C18(100 mm×2.1 mm,1.8 μm)色谱柱,以0.2%甲酸溶液-乙腈(90∶10)为流动相,流速为0.3 mL/min,柱温为30 ℃,进样量为5 µL。
2.2 质谱条件
电子喷雾离子源(ESI)正离子模式,喷雾电压4 500 V;锥孔电压20 V;离子源电压50 V;离子源温度150 ℃;雾化气压力7.00 bar;锥孔气流量150 L/Hr;喷雾气流量1 000 L/Hr;碰撞能量10~40 eV;采用全扫描模式,扫描质量数范围50~500 m/z。
2.3 样品制备
2.3.1 供试品溶液的制备精密称取头孢拉定胶囊内容物25 mg,置于25 mL 容量瓶中,加水溶解并稀释至刻度,摇匀,滤过,即得。
2.3.2 对照品溶液的制备精密称取头孢拉定对照10.25 mg,置100 mL 量瓶中,加水溶解并稀释至刻度,摇匀,滤过,即得。
2.3.3 强制降解试验溶液的制备强制降解条件如下:酸破坏(1 mol/L HCl 1 mL,50 ℃水浴,1 h);碱破坏(0.05 mol/L NaOH 1 mL,50 ℃水浴,3 h);氧化破坏(0.5% H2O21 mL,室温,1 h)。酸破坏和碱破坏在终止破坏前用相应浓度的NaOH 溶液和HCl 溶液中和,所有破坏样品最终用水稀释成含头孢拉定约1 mg/mL 的强制降解试验溶液。
2.4 结果
2.4.1 强制降解试验结果强制降解试验结果表明:在酸、碱和氧化试验均产生相应杂质,三种条件下杂质种类和数量有区别,尤其是在氧化试验条件和碱强制降解条件下区别明显,酸强制降解条件下产生的杂质与氧化试验条件下产生的杂质相似。随着强制降解条件的加强,杂质的离子流丰度越来越强。在供试品中除主峰外还检出了杂质12 头孢氨苄和相对保留时间为1.67 的杂质11,质荷比352 m/z 推测为4'-5'-二氢头孢拉定,且该杂质峰在碱强制降解条件下容易被降解。

图2 总离子流图(50~500m/z):a 氧化试验;
2.4.2 杂质结构的分析首先根据一级质谱扫描得到各杂质的母离子,确定其相对分子质量和元素信息,再根据二级质谱获得碎片信息,结合化合物的一般裂解规律,推测其可能的化学结构。
杂质1 相对保留时间为0.1,氧化试验产生,一级质谱图中[M+H]+离子峰为416 m/z,与[C16H21N3O8S]+离子式相对应,二级质谱扫描的离子碎片峰399、273、158、140 m/z 等,均可由此结构经合理裂解产生。推测裂解过程及二级质谱图见图3。
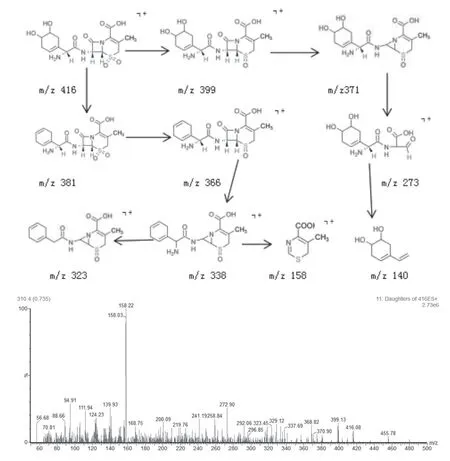
图3 416 m/z结构裂解过程图(a)和二级质谱图(b)

表1 强降解产物的LC-MS/MS信息
杂质2 和杂质3 相对保留时间分别为0.12 和0.16,在酸降解,氧化试验中均有产生,其质谱图基本相同,[M+H]+离子峰为366.24 m/z,[M+Na]+离子峰为离子388.11 m/z,与[C16H19N3O5S]+离子式相对应。子离子峰348 m/z,为[M+H]+离子脱去一分子H2O 后形成的碎片离子,说明结构中含有裸露的羟基。子离子峰322 m/z 与[M+H]+离子相差44,推测为[M+H]+离子脱去一个羧基,说明结构中含有羧基,综上所述,见图4a(为《欧洲药典》杂质C 和D 异构体)和图4b,质谱图见图4c。
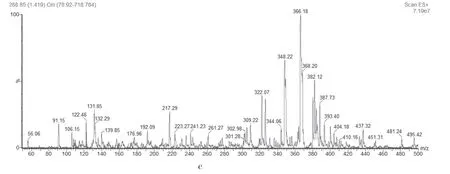
图4 杂质2和3的结构图(a、b;368 m/z)和质谱图(c)
杂质4、杂质5 和杂质6 相对保留时间分别为0.33、0.39 和1.11,在酸、碱降解和氧化试验中均有产生。其[M+H]+离子峰均为368 m/z,质谱图相似,与[C16H21N3O5S]+离子式相对应,可能为同分异构体。有文献报道[1]推测为β-内酰胺环上的酰胺键断裂,被氧化水解形成羧基。在氧化试验中还产生了杂质7 相对保留时间为0.2,其[M+H]+离子峰也为368 m/z,其离子碎片与杂质4、5 和6 的子离子碎片相似,但有一定差别,其结构有待进一步考察,结构见图5。


图5 368 m/z结构图
杂质8 相对保留时间为0.58,在氧化试验中产生。在质谱图中存在340 m/z 和363 m/z 的离子峰,分别推测为[M+H]+和[M+Na]+峰,即杂质8相对分子质量为339。其二级质谱子离子碎片189、122、322、278、305 m/z,均可由此结构经合理裂解产生。推测裂解过程及质谱图,见图6 和图7。

图6 杂质8裂解过程图

图7 杂质8质谱图
杂质9 和杂质10 相对保留时间分别为0.26和0.45,均在碱降解试验中产生。杂质9 质谱图中[M+H]+离子峰为177m/z,子离子峰149m/z为[M+H]+脱去一个羰基[4],质谱图及结构推测如图8。杂质10[M+H]+离子峰为220 m/z,其结构有待进一步考察。
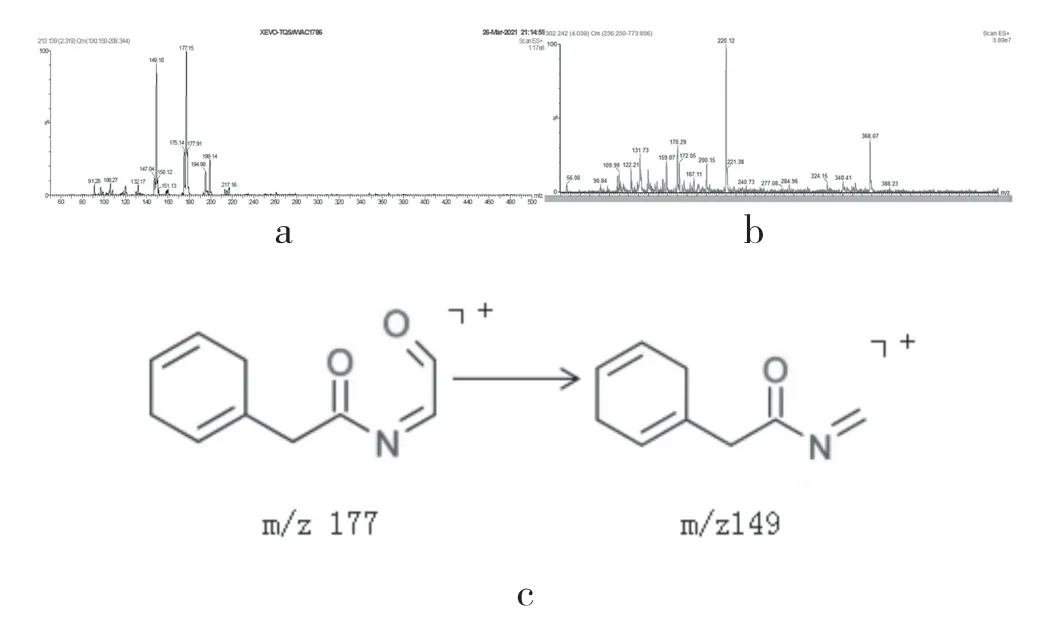
图8 杂质9和10的质谱图(a,b)及杂质9(c)结构推测
3 讨论
3.1 色谱条件选择与优化
据报道,对本品进行杂质检查时大多采用磷酸盐缓冲液作为流动相,不适合电喷雾离子化的质谱分析。本研究建立了适合头孢拉定LC-MS/MS 分析的色谱条件,并对其进行了优化。通过对甲醇:水、甲醇:0.1%甲酸水、乙腈:水、乙腈:0.05%甲酸水、乙腈:0.1%甲酸水、乙腈:0.2%甲酸水、乙腈:醋酸铵缓冲溶液和甲醇:醋酸铵缓冲溶液等8 种流动相系统,分别调节不同比例进行试验,结果表明在“2.1”项流动相条件下,头孢拉定峰保留时间适当,杂质有效分离,且峰型好,是较理想的流动相。
3.2 头孢拉定强降解杂质
本研究采用LC-MS/MS 分析法,检测出头孢拉定胶囊强制降解产物12 个,根据质谱分析和相关文献报道,其中可能包含了《欧洲药典》收载的杂质C 和杂质D,以及头孢氨苄和4'-5'-二氢头孢拉定等已知杂质。其余为未知杂质,其中m/z368 的杂质有4 个,有文献报道其1 种相关结构,针对这4 种杂质是同分异构体还是不同物质,有待进一步考察。其余的未知杂质结构未见相关文献报道。利用二级质谱进行结构解析,推测出其可能的结构,但结构的精准鉴定还有待借助多种手段进一步确证。
