亨廷顿病临床研究及家系分析
2021-07-07王银萍刘恒方张敏崔明
王银萍,刘恒方,张敏,崔明
(郑州大学第五附属医院 神经内三科,河南 郑州 450000)
亨廷顿病(Huntington’s disease,HD)又称为亨廷顿舞蹈病,是一种罕见的神经退行性疾病,在亚洲人群中发病率较低[1]。该病以常染色体显性遗传方式遗传,是由于4p16.3区域的HTT基因(IT15)中的CAG重复序列异常扩增[2],引起亨廷顿蛋白功能异常,从而导致特定神经元细胞死亡的一种疾病,主要发生在大脑纹状体和皮质区域[3]。HD典型表现为舞蹈样不自主运动、精神行为和认知功能改变,平均发病年龄30~50岁,平均病程17~20 a。另外,约5%的病例在儿童或青少年时期发病,称为青少年HD,主要症状为认知障碍、癫痫发作、共济失调、肌张力以及精神行为改变[4]。本研究中收集了一个HD家系完整的临床资料,包括家庭环境、临床症状、影像检查、脑电图、基因检测、治疗方法及疗效等,记录患者病情进展以及绘制完整的遗传家系图,总结HD临床特点及遗传规律以提升对疾病的整体认知,识别症状前患者,早诊断早治疗,提高生活质量,并提供生育指导。
1 资料与方法
1.1 一般资料先证者为郑州大学第五附属医院神经内三科于2017年8月收治的1例初步诊断为HD的30岁汉族女性。据先证者描述,该家系四代6例已发病患者长期生活于同一个小区,在知情、自愿的原则下,本研究抽取了先证者及其2个儿子的外周血进行HD基因测试。
1.2 研究方法对此HD家系进行完整的病史采集,包括详细的神经系统检查,评估认知功能及精神状态,并绘制家系图。完善相关辅助检查:(1)头颅核磁检查;(2)脑电图检查;(3)从受检者外周血中提取DNA,采取荧光标记引物扩增HTT基因,进行片段分析。采用亨廷顿舞蹈病统一评定量表(Unified Huntington’s Disease Rating Scale,UHDRS)评定HD患者在运动、认知、精神行为、日常生活能力等方面的表现,从而衡量HD患者病情进展情况[5]。结合该家系临床特点及遗传规律,对该家系进行遗传学指导。
2 结果
2.1 家系分析该家系4代33人截至目前共出现6例患者,男3例,女3例,发病年龄8~58岁,病程5~24 a,其中5例患者已死亡(图1)。第一代发病年龄58岁,病程17 a;第二代患者发病年龄30、45岁,中位数为37.5岁,病程7、24 a,中位数为15.5 a;第三代患者发病年龄8~28岁,中位数为26岁,病程5~23 a,中位数为7 a。该家系遗传病符合常染色体显性遗传规律,同时存在遗传早现情况,且下一代生存时间更短。
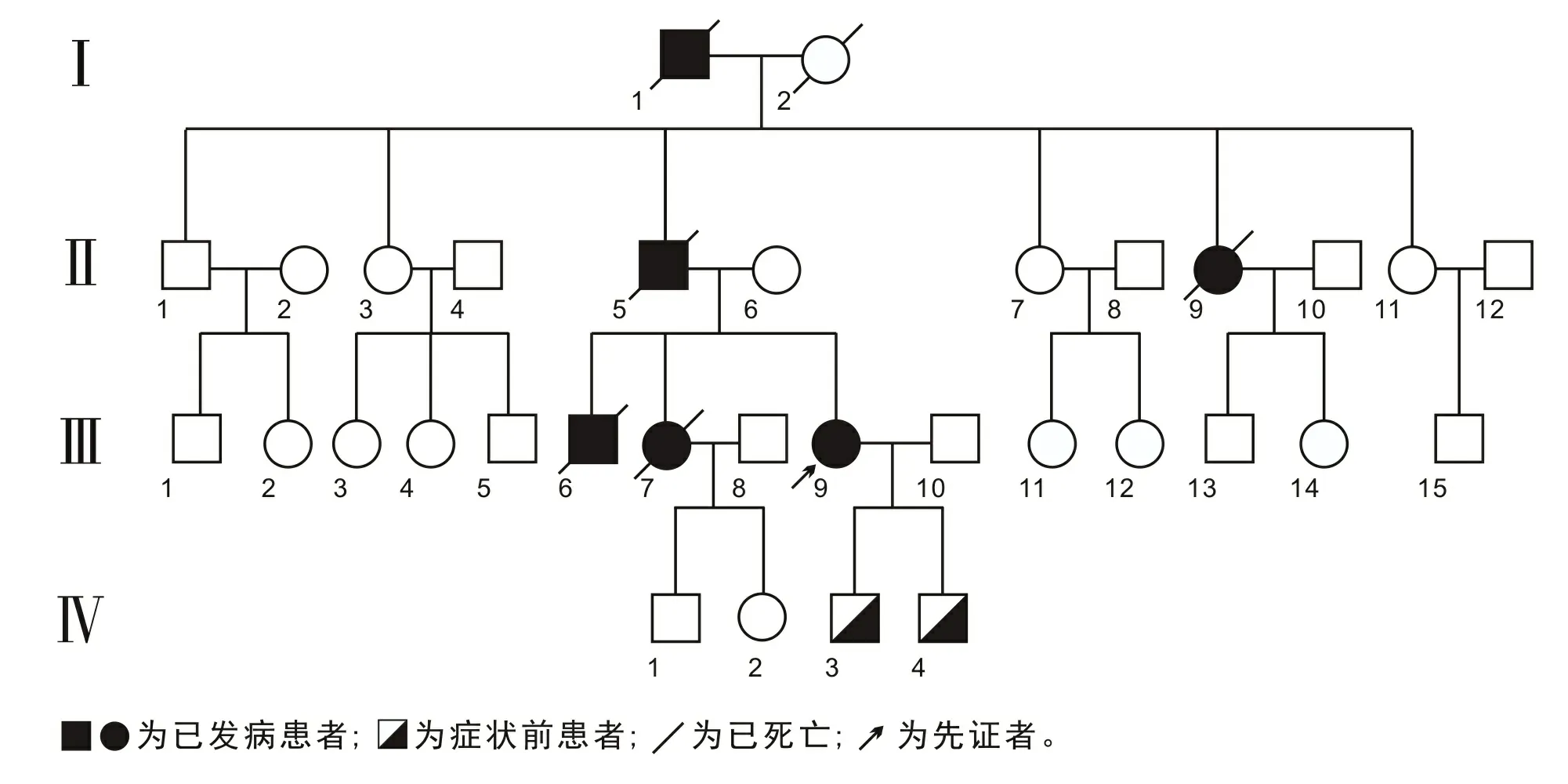
图1 遗传家系图
2.2 临床资料6例患者均表现为四肢及躯干不自主舞动,行走不稳,运动迟缓。先证者(Ⅲ9)33岁,28岁发病,表现为四肢间断不自主舞动,动作迟缓,症状逐渐加重,30岁入院时表现为全身不自主舞动,面部表情怪异,舞蹈样步态,运动迟缓,反应迟钝,注意力减退,记忆力下降。神经查体:言语清,眼球活动正常,伸舌可,肌力正常,肌张力低,四肢腱反射活跃,双侧巴氏征可疑阳性,共济尚可,感觉及自主神经系统检查未见明显异常。先证者祖父(Ⅰ1),58岁发病,运动障碍症状较轻,易躁,认知功能改变不明显,75岁去世,病程17 a;先证者父亲(Ⅱ5),30岁发病,抑郁,37岁因自戕去世,病程7 a;先证者姑姑(Ⅱ9),45岁发病,69岁去世,病程24 a;先证者哥哥(Ⅲ6),8岁出现痫性发作,智力低下,23岁出现四肢舞蹈样抖动,食欲旺盛,体质量下降,31岁去世,病程23 a;先证者姐姐(Ⅲ7),26岁发病,易躁易怒,入睡困难,食欲增加,后期极度消瘦,33岁去世,病程7 a。
该家系中5例患者(Ⅰ1,Ⅱ5,Ⅱ9,Ⅲ7,Ⅲ9)早期均出现典型的舞蹈样动作、行走不稳,1例患者(Ⅲ6)儿童时期出现癫痫发作,智力低下,行走不稳,15 a后出现舞蹈样抖动。5例患者(Ⅰ1,Ⅱ5,Ⅱ9,Ⅲ7,Ⅲ9)运动症状出现前已有精神行为学改变,3例患者(Ⅰ1,Ⅱ9,Ⅲ7)表现为易躁易怒,1例患者(Ⅱ5)出现明显抑郁,1例患者(Ⅲ9)表现为情感淡漠及缺少主动性,且疾病晚期均表现为抑郁倾向、刻板。6例患者均出现入睡困难、睡眠中多动等症状,4例患者(Ⅱ9,Ⅲ6,Ⅲ7,Ⅲ9)食欲异常增加,4例患者(Ⅰ1,Ⅱ9,Ⅲ6,Ⅲ7)出现体质量明显下降。见表1。
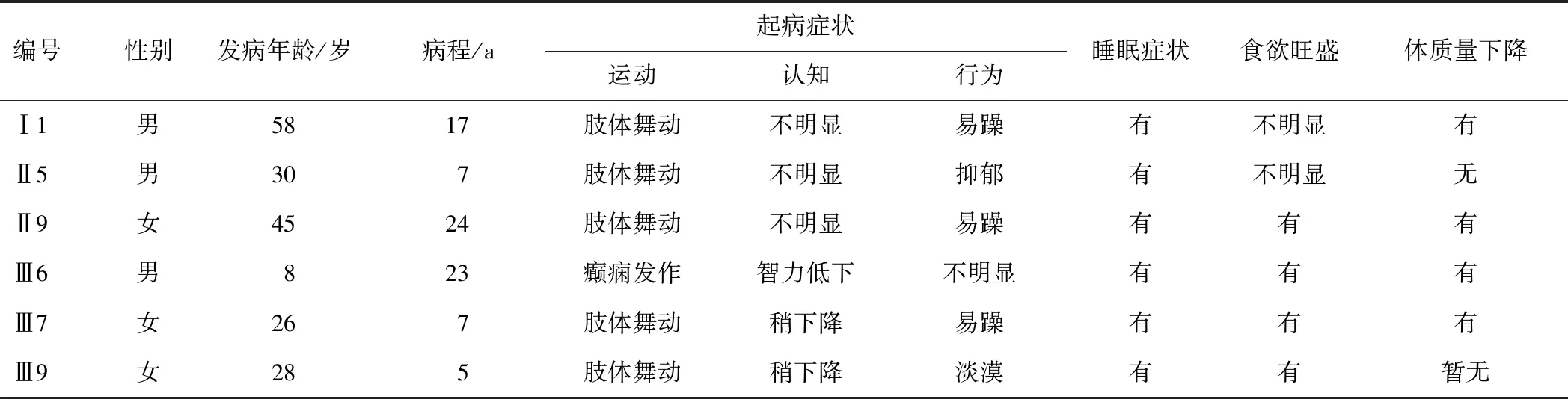
表1 该家系患者的临床资料
2.3 辅助检查结果
2.3.1头颅核磁结果 先证者Ⅲ9尾状核稍萎缩,脑室系统稍扩大,大脑皮质轻度萎缩,脑沟、脑裂稍增宽、加深(图2);患者Ⅲ7发病时查头颅核磁未见明显异常。前驱期患者Ⅳ3、Ⅳ4头颅核磁无异常。
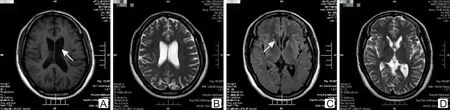
注:A、B提示侧脑室稍扩大,大脑皮质轻度萎缩,脑沟、脑裂稍增宽;C、D可见状核头稍萎缩。图2 先证者Ⅲ9头颅核磁结果
2.3.2脑电图检查结果 先证者脑电图检查结果提示其安静闭目时未见明显双侧后头部α波;全导波幅较低,前额部可见少量5~7 Hz的θ波。见图3。

图3 先证者脑电图结果
2.3.3HD基因测试 由于家庭原因,该家系目前仅先证者及其两个儿子进行了HD基因测试,检测发现先证者HTT基因(CAG)拷贝数分别为18和53;大儿子(Ⅳ3,12岁)HTT基因(CAG)拷贝数为19和53次,小儿子(Ⅳ4,6岁)HTT基因(CAG)拷贝数为19和58次。3人均符合HD分子遗传诊断特征。见图4~6。
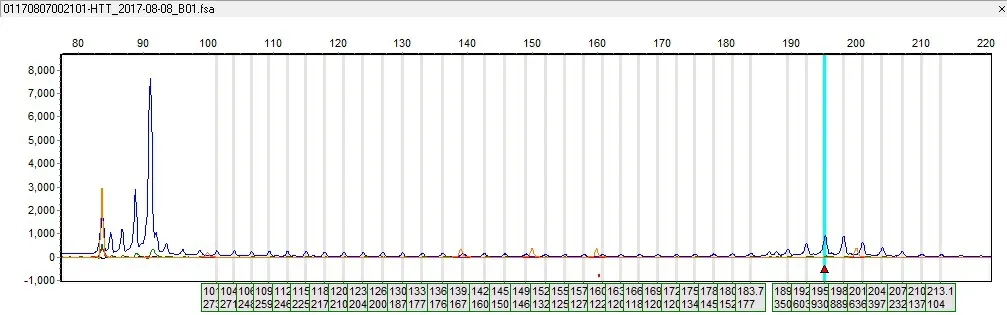
图4 先证者(Ⅲ9)的HTT基因检测结果

图5 大儿子(Ⅳ3)的HTT基因检测结果
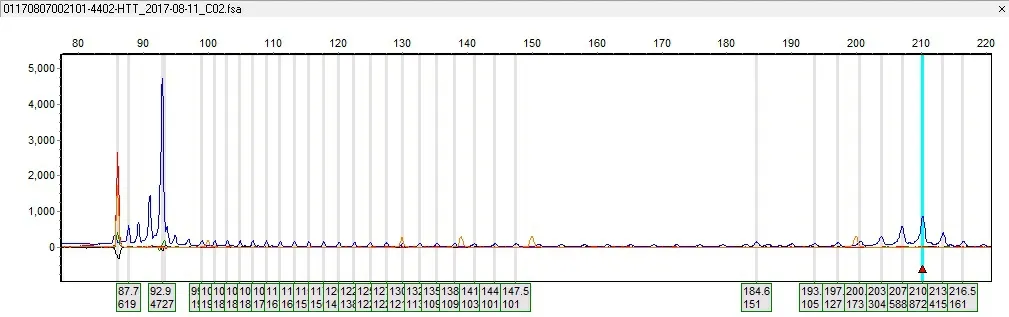
图6 小儿子(Ⅳ4)的HTT基因检测结果
2.4 治疗与疾病进展患者出院后规律口服氟哌啶醇,症状仍逐渐加重,目前先证者主要出现全身不自主舞动、行动迟缓、走路不稳、痴呆、少动、冷漠、言语不清、食欲增加、入睡困难及睡眠中多动等现象。UHDRS量表评分显示,目前,患者运动总分、行为评分得分增加,总功能评分、独立量表评分、日常任务清单评分、Stroops试验得分下降。见表2。

表2 先证者3 a的UHDRS评分对照表(分)
3 讨论
HD患者多隐匿起病,起病症状多样,该家系1例青少年HD患者早期以癫痫发作起病,其余5名患者早期出现不自主的舞蹈样动作,并且不自主抖动从肢体远侧开始,程度较小,随着病程进展累及近端肢体、全身包括面部,幅度更大,且易跌倒,在疾病的晚期,运动症状倾向于运动迟缓甚至肌肉强直。既往国内研究发现50%以上患者在运动症状出现前已有行为学改变[6],国外研究发现HD中,抑郁情绪的发病率为33%~69%[7],可能与焦虑、易怒和冷漠的发病率相当。该家系5/6例患者运动症状前已出现精神行为异常,并且早期以易躁、冲动和情绪调节不良为主,后期均有不同程度的冷漠、丧失主动性、创造力和好奇心。国外研究报道HD认知功能改变首先是记忆功能受损[8],该家系患者早期出现的认知功能改变首先表现在注意力、执行能力下降,随着疾病进展出现记忆检索障碍,部分患者最终演变为皮质下痴呆。6例患者均出现睡眠紊乱,大多表现为入眠困难,睡眠中多动,可能与下丘脑病变有关[9-10]。体质量已被证明是临床HD进展的预测因子之一[11]。国外报道体质量下降可能与吞咽障碍、胃肠道功能减退、伴食欲减退的抑郁症、不自主运动有关[12],该家系部分患者出现食欲异常增加,但早期体质量下降的现象,考虑与异常亨廷顿蛋白造成全身高代谢状态[13]更为相关。
据报道HD患者发病年龄与CAG重复次数、环境和遗传因素有关[14]。该家系发病年龄在连续的世代中降低,存在遗传早现现象,与既往报道相符。遗传早现大多是由于父系遗传过程中CAG重复序列扩增导致[15],但该家系母系遗传时(Ⅲ9与Ⅳ4)也出现了CAG重复序列扩增的现象。
先证者存在典型的舞蹈样症状,头颅核磁提示轻度脑萎缩,基因检测提示HTT基因CAG异常重复次数为53次,依据HD的诊断与治疗指南(2011)[16],可确诊为HD患者。Ⅲ6患者早期表现为癫痫发作,智力低下,数年后出现运动症状,依据临床症状及阳性家族史可诊断为HD患者。先证者2个儿子为症状前患者,经基因检测确诊,但尚未发现异常症状,需继续随访,尽早干预以减缓临床进展,并可在生育期对患者提供产前诊断[17],以降低子代发病率。尽管HD早期症状多样,目前舞蹈样动作仍是HD诊断的关键,阳性家族史的患者依据典型临床症状、神经系统查体即可临床确诊,阴性家族史的患者主要依靠基因检查确诊诊断,正常人HTT基因CAG重复次数不超过26,CAG重复次数大于40对此病具有诊断意义[18],头颅影像学尤其脑功能成像检查结果可协助诊断。另外,临床确诊HD还需注意与小舞蹈病、迟发性运动障碍等疾病[19]相鉴别。
虽然我国对HD的了解逐渐增加,但尚未找到有效缓解疾病进展的方法,临床治疗多为对症治疗,川芎嗪是唯一有证据表明可以治疗HD的药物,特别是减轻舞蹈样症状,提高患者生活质量[20]。现阶段许多新型治疗方案也在开发研究中,旨在减缓疾病进展。这些方法中,基于DNA和RNA靶向治疗从而降低异常亨廷顿蛋白水平及生物功能的疗法具有很大的治疗前景,包括常见的反义寡核苷酸、RNA干扰、小分子剪接调节剂和锌指蛋白转录因子应用[21-23],为HD患者的治疗带来了希望。
将来HD的治疗可能会给HD患者及其家属的生活带来前所未有的改善,但对这些疗法的评估需要新的工具,以实现更快、更有效的评估。先证者UHDRS评分提示运动功能、认知功能、日常生活能力明显下降,可以直观地显示出疾病的纵向进展,可以用来评价疾病的治疗效果,同时可以用于比较不同患者的临床症状。相关研究表明脑脊液异常亨廷顿蛋白、血浆神经纤维丝轻链含量也可能成为疾病进展情况的标志物[24]。另外MRI的尾状核、白质、全脑体积,核磁弥散张量成像测量的脑白质完整性,PET呈现的脑代谢情况等影像学手段也与疾病进展相关[25-26]。这些生物标志物的发现将使更早识别HD患者、更有效地评估疾病进展成为可能。
随着对HD发病机制认识的不断深入,降低致病性亨廷顿蛋白水平及其生物学效应的治疗策略也不断进入临床试验阶段,HD患者及家人应以积极、乐观的心态面对疾病,关注最新研究进展,科学生育,提高生存质量。
