关于阿达木单抗生物类似药质量相似性评价要点的初步探讨
2021-05-19程速远赵靖胡莹莹白玉
程速远,赵靖,胡莹莹,白玉
国家药品监督管理局药品审评中心,北京100022
近年,生物类似药研发的热度日增,已有近200个单抗生物类似药候选药进入临床研究阶段,其中阿达木单抗(adamumab)是申报量最高的生物类似药之一。截至2020年7月,我国已有2种阿达木单抗生物类似药上市[1],另有2个厂家产品进入申报生产阶段,22个厂家产品处于临床试验阶段。美国FDA已批准安进公司、勃林格殷格翰药业有限公司、诺华-山德士公司、三星Bioepis/默沙东集团、辉瑞制药有限公司和迈兰公司/协和发酵株式会社6家公司研发的阿达木单抗生物类似药上市,欧盟也已批准上述除辉瑞公司之外5家公司研发的阿达木单抗生物类似药上市。
本文通过汇总与比较原研厂及国内26家阿达木单抗生物类似药研发企业的表征分析数据,结合各国生物类似药相关指导原则及参考文献,对阿达木单抗生物类似药质量相似性的技术评价要点进行初步探讨。
1 阿达木单抗质量相似性评价
阿达木单抗(商品名Humira®)系由美国雅培公司开发,采用中国仓鼠卵巢细胞系表达制备的首个重组全人源化IgG1κ型单克隆抗体,由2条重链和2条轻链组成,共含1 330个氨基酸,每条重链Fc部分相同糖基化位点含有典型的N-端连接糖链。阿达木单抗通过特异性结合可溶性肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α),阻断其与细胞表面受体p55和p75相互作用,从而有效抑制TNF-α诱导的多种病理效应。另外,阿达木单抗还可与跨膜TNF-α结合,通过Fcγ受体结合的抗体依赖细胞介导的细胞毒作用(antibody-dependent cell-mediated cytotoxicity,ADCC)和补体依赖的细胞毒作用(complement dependent cytotoxicity,CDC),诱导细胞凋亡等效应,清除部分致病靶细胞。
生物类似药的研发是以原研药的关键质量属性为目标,以证实候选药物与原研药的质量相似性或二者结构功能差异不会引起安全性或有效性方面具有临床意义的差异作为生物类似药药学开发与评价的核心[2]。在生物类似药的开发过程中,一方面应关注原研药上市后变更,尽可能收集较多批次的原研参照药;一方面应关注使用统计方法的策略和相关法规随着经验积累进行的调整[3]。
1.1 阿达木单抗原研产品注册与工艺变更情况 阿达木单抗于2002年在美国注册上市,2003年在欧盟注册上市,现已在90多个国家和地区上市,批准用于包括类风湿性关节炎(rheumatoid arthritis)、银屑病关节炎(psoriatic arthritis)、强直性脊柱炎(anky-losing spondylitis)、克罗恩病(Crohn disease)、斑块银屑病(plaque psoriasis)和溃疡性结肠炎(ulcerative colitis)、幼年特发性关节炎(juvenile idiopathic arth-ritis)、儿童克罗恩病(Crohn disease pediatric)、特定类型葡萄膜炎(specific types of UVEITIS)、治疗影像学阴性中轴脊柱炎(nr-axSpA)等多种适应证。阿达木单抗于2010年在中国上市,先后获批用于类风湿性关节炎、强直性脊柱炎和银屑病3种适应证,并于2020年获批新增克罗恩病、幼年特发性关节炎、儿童斑块银屑病和葡萄膜炎4种适应证。
根据欧洲药监机构公布的公众评估报告(European Public Assessment Report,EPAR)信息,阿达木单抗自2002年上市后至少经过28次不同风险等级的工艺变更,包括8次低风险变更,17次中度风险变更和3次重大风险变更[4]。从阿达木单抗原研公司在我国药品再注册和补充申请等受理情况分析,中国已上市的阿达木单抗在原液生产场地、生产工艺、包材、包装规格、制剂处方等方面的确存在多次工艺变更。上述工艺变更可能导致阿达木单抗的质量在其产品生命周期内发生“漂移”。
1.2 生物类似药质量相似性评价的一般考虑 生物制品具有分子量大、结构复杂、生物活性对其结构完整性依赖性强、变异体多、生产工艺复杂等特点[5],因此,随着国内外生物类似药研发和评价的不断深入,生物类似药相似性评价标准的推出尤为重要。我国于2015年发布了《生物类似药研发与评价技术指导原则(试行)》,明确了相似性评价的基本原则,包括比对原则、逐步递进原则、一致性原则和相似性评价原则[6]。
通过借鉴国际相关法规政策和近年的审评经验积累,国家药品监督管理局药品审评中心于2020年8月发布了《生物类似药相似性评价和适应证外推技术指导原则(征求意见稿)》,提出相似性是指生物类似药与参照药之间高度相似,在纯度、安全性及有效性不存在有临床意义的差别。进一步明确了应基于对参照药质量属性的认知程度及其与临床风险获益的相关性,对质量属性进行高、中、低风险的分级。高、中风险质量属性可采用验收标准范围方法进行定量评估;低风险和无法采用定量方法评价的质量属性可采用头对头定性比对或图谱比对的方法进行评估。用于定量评估的质量范围通常定义为μRXσR/μR+XσR,其中μR为检测样本的平均值,σR为标准偏差,系数X的设定需根据质量属性的风险等级和方法的变异性等进行科学论证,针对高风险质量属性应合理收紧系数。根据质量属性权重和相似性评价目标,也可采用其他统计方法进行数据分析,如等效性检验。鼓励采用先进的、灵敏度高的技术和方法对候选药和参照药开展全面质量比对研究。对候选药与参照药存在的与临床风险获益相关性认知尚不充分的差异,应设计针对性的比对试验研究,以证实质量差异的不确定性对药物安全性、有效性和免疫原性等方面的影响。必要时,还需要提供额外的非临床和/或临床证据,科学地论证质量差异是否具有临床意义[7]。
1.3 关键质量属性的识别 阿达木单抗属于典型的IgG1型单克隆抗体,糖基化修饰复杂,具有多种质量属性,根据对其生物学活性、药代动力学(pharmacokinetic,PK)、安全性和免疫原性的潜在影响及对应的临床用药风险获益程度,确定阿达木单抗的关键质量属性并对质量属性进行风险等级排序,见表1[8]。
1.4 关键质量属性相似性评价
汇总26家国内阿达木单抗生物类似药厂家(共109批,其中37批重复)、原研药厂家(30批)、中国食品药品检定研究院(简称中检院)质量复核(9批)的质量研究数据,并结合文献[2,9-10]对阿达木单抗原研药的关键质量属性检测数据,统计后确认初步质量相似性评价标准(Mean±3SD),再评估各家候选药的各质量属性检测结果是否符合该相似性评价标准范围。
1.4.1 结构确证
1.4.1.1 氨基酸序列 生物类似药研发的前提是氨基酸序列的一致性,通常多种酶切后采用液相色谱-质谱联用(简称液质联用)的方法进行肽图检测,大部分阿达木单抗生物类似药候选药的二级质谱覆盖率可达100%,同时应证明氨基酸序列与参照药一致。
1.4.1.2 分子量 单抗分子量包括完整分子量、还原和去糖后重链及轻链分子量。完整分子量通常采用点喷射离子化飞行时间质谱进行检测;还原态的去糖基化重链和轻链的质量分析则多采用多肽-N-糖苷酶(PNGase F)去除N连接的糖链后变性还原,再进行液质联用分析。各厂家的阿达木生物类似药候选药切糖分子量与参照药一致性良好,通常切糖后重链和轻链分子量分别为49 200和23 412,与理论值一致。完整蛋白水平的分析难以提供糖基化修饰的精细信息,在质谱法测定中易受蛋白同位素峰的影响,仅可提供抗体的平均分子量信息。鉴于目前收集到的各家完整蛋白分子量批次有限,暂未纳入统计。但候选药厂家应尽可能采用高分辨率质谱与参照药开展头对头的检测,并分析各分子量与参照药的异同。
1.4.1.3 二硫键及自由巯基 通过比较还原条件和非还原条件下的肽图,可鉴别出含有二硫键的多肽,进一步确认二硫键的结构。阿达木单抗共有16对二硫键,包含12对链内二硫键及4对链间二硫键,理论上不应存在游离巯基。对各厂家二硫键鉴定结果均与理论一致,自由巯基含量一致性良好,均保持在极低水平。
1.4.1.4 糖基化修饰 通常采用PNGase F酶切糖链,经糖链荧光标记后通过液质联用进行糖型分析。阿达木单抗的糖基化位点位于重链第301位天冬酰胺(Asn)上,主要糖型为岩藻糖基化二天线寡糖(如G0F、G1F、G2F等),占糖型总量的82.3%~95.1%,其中G0F占74.3%~76.5%,G1F+G2F占13.3%~24.9%[9],其他糖型还有G0、高甘露寡糖(如Man5、Man6等)、半乳糖基化、唾液酸化等。其中,半乳糖基化基团包括全部复合物及至少含1个末端半乳糖的杂合聚糖结构,半乳糖基化对补体C1q与IgG1的结合较重要,是通过CDC激活IgG1介导细胞裂解的关键步骤;高甘露糖聚糖类型是人血清IgGs中天然存在的,可通过结合至甘露糖结合受体引起清除率变化而影响PK,并通过结合FcγRⅢa影响ADCC活性,如Fc端N-连接聚糖缺少核心岩藻糖可显著提高IgG1结合至FcγRⅢa的亲和力,从而提高ADCC活性;唾液酸含量会影响PK和有效性[11],但阿达木单抗的唾液酸含量较低,对产品功能相似性影响较小。另外,阿达木单抗具有一定的ADCC和CDC效应,与跨膜TNF-α作用机制相关的克罗恩病和葡萄膜炎可能与此有关,在适应证外推中应考虑影响ADCC和CDC效应的糖基化水平。鉴于糖基化与阿达木单抗的生物活性、有效性、安全性及PK等密切相关,不同生物类似药应与原研药具有相似的糖型分布。
各生物类似药厂家获得的阿达木单抗主要糖型的分布较为相似,但由于各家对糖型的分类、采用的检测方法及积分计算方法不同,原研厂家自测和生物类似药厂家测定的参照药糖型含量区间范围存在一定差异,个别候选药的部分糖型含量超出了初步拟定的相似性标准范围。部分候选药主要糖型含量见图1。
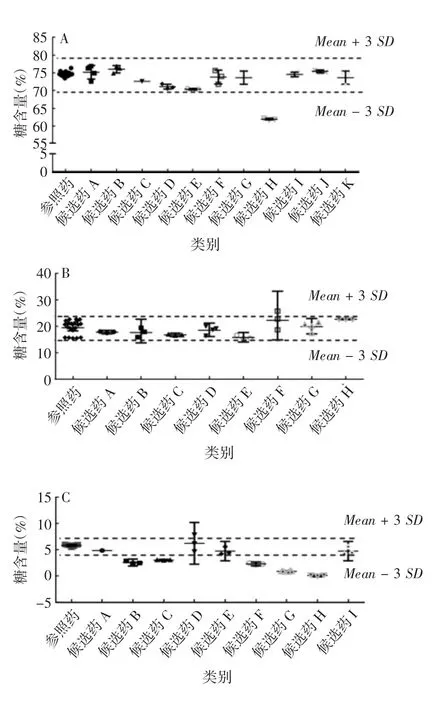
图1 阿达木单抗参照药和候选药的糖型含量
1.4.1.5 高级结构 一般采用圆二色谱法、荧光光谱法及差示扫描量热法等进行单克隆抗体高级结构的检测,候选药应与参照药进行头对头分析比较,检测图谱、吸收峰和Tm值应与参照药一致。
1.4.1.6 等电点 等电点受蛋白质氨基酸序列和高级结构的影响,一般采用毛细管等电聚焦(capillary isoelectric focusing,cIEF)法检测等电点。阿达木单抗为糖基化蛋白,等电点的图谱为一组峰,各峰的出峰时间与峰高应与原研药一致。各生物类似药厂家的主峰出峰时间与参照药基本一致,但酸性峰和碱性峰分布存在一定差异。
1.4.2 生物活性
阿达木单抗功能相似性评价包括与可溶性TNF-α的结合动力学及与跨膜TNF-α相对结合力的检测,同时还应检测TNF-α细胞毒中和作用及与Fcγ受体(FcRⅠa、FcRⅡa、FcRⅢa)和新生儿受体(FcRn)的结合能力,并分析其ADCC及CDC活性。通常采用ELISA法测定与可溶性TNF-α的相对结合力,细胞凋亡抑制法检测细胞活性,表面等离子体共振法(surface plasmon resonance,SPR)测定样品和TNF-α和Fc受体的亲和力。
1.4.2.1 结合活性 采用ELISA法检测样品与可溶性TNF-α的结合能力,一般拟定质量标准为对照品的75%~125%。
1.4.2.2 细胞活性TNF-α可诱导L-929小鼠成纤维细胞凋亡,TNF-α与阿达木单抗特异性结合后,阻断了其对靶细胞的凋亡作用,中和了其对L-929细胞毒性。采用TNF-α细胞毒中和法,通过比较参比品和供试品的EC50值,计算供试品的凋亡抑制生物活性。一般拟定质量标准范围为对照品的80%~125%,由于方法的变异性较大,部分生物类似药企业拟定质量标准为70%~150%。鉴于候选药质量标准应不低于参照药,建议通过方法优化提高方法的精密度,尽量收紧至原研药的质量标准范围。
1.4.2.3 Fc受体及C1q亲和力 抗体Fc端与免疫细胞表达的Fc受体(FcRⅠa、FcRⅡa、FcRⅢa)、新生儿受体(FcRn)及血液中的补体(C1q)的结合对抗体的PK有一定影响,因此需要关注候选药与参照药在样品Fc端多种受体亲和力方面的相似性。Fc受体及C1q亲和力通常采用SPR法检测,方法的变异性使各家检测结果差异较大,因此在相同检测条件下的头对头研究十分必要。
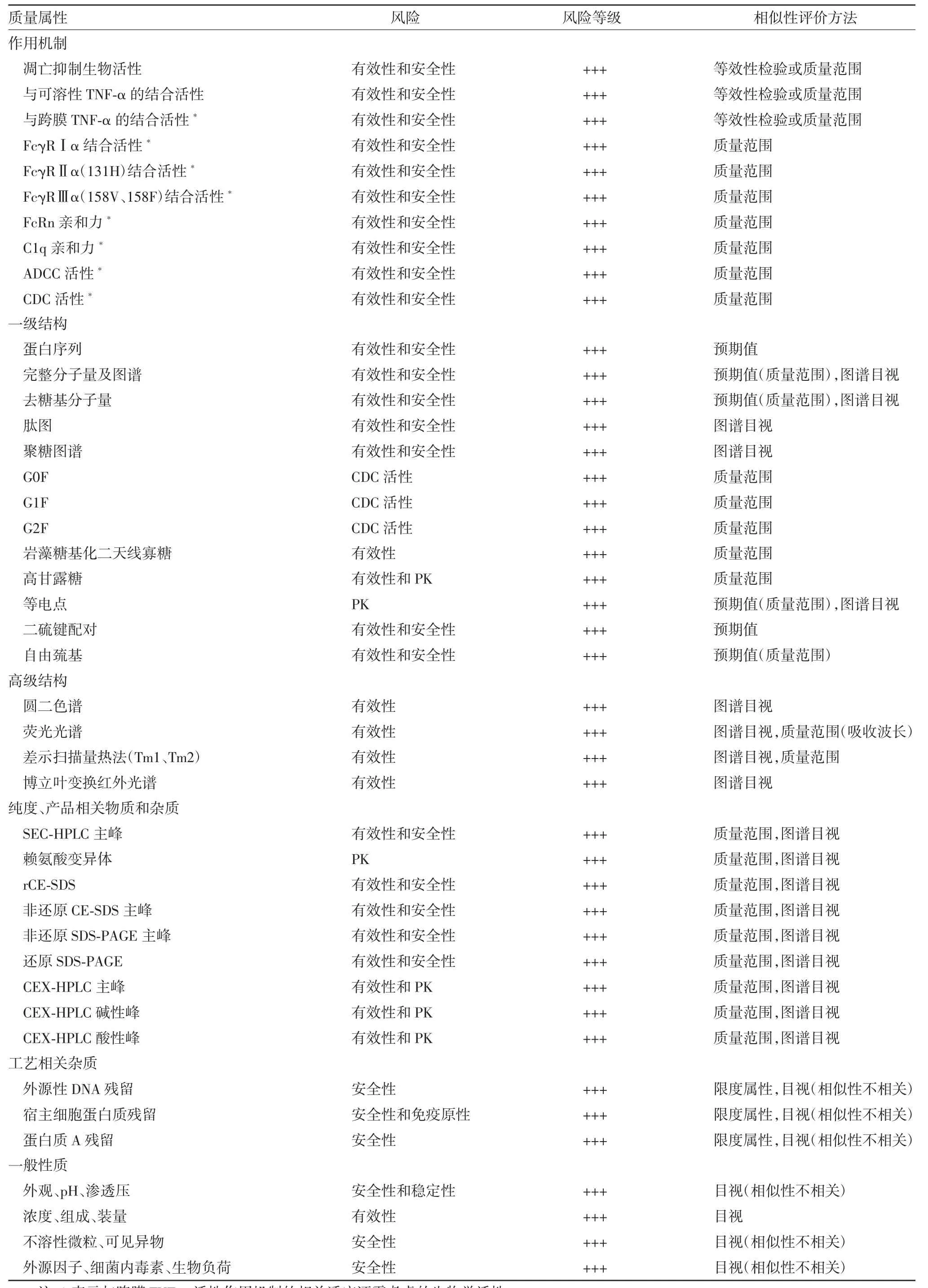
表1 阿达木单抗关键质量属性
1.4.2.4 ADCC和CDC活性 通常阿达木单抗与可溶性TNF-a结合不具有ADCC和CDC活性,但与跨膜TNF-α结合后可发挥ADCC和CDC活性。ADCC检测一般采用报告基因检测系统,靶细胞为细胞表面表达TNF-α的CHO-K1/hmTNF-α细胞,阿达木单抗可与细胞膜表面TNF-α结合,启动效应细胞报告基因表达,促进发光底物发光,发光程度与抗体浓度呈正相关。CDC通过阿达木单抗与CHOK1/hmTNF-α细胞表面表达的TNF-α结合,形成复合物激活补体经典途径,所产生的攻膜复合物对靶细胞发挥裂解作用,通过乳酸脱氢酶(lactatedehy-drogenase,LDH)及钙黄绿素(calcein AM)的释放来检测CDC作用,其显色程度与抗体浓度呈正相关。由于各厂家检测生物活性时均采用自制对照品作为标准品,横向比较和统计各厂家生物活性检测结果意义较小。统一国家标准品的建立将有助于同类产品生物活性等质量的平行对比。
1.4.3 纯度
1.4.3.1 SEC-HPLC纯度 通常采用SEC-HPLC法检测分子大小异构体,主峰前后分别是聚体和降解片段。聚体的存在会带来免疫原性风险,并影响PK;降解片段可能会造成结合活性下降,对有效性产生影响。各生物类似药厂家测得的阿达木单抗单体纯度范围为98.5%~100.6%,个别候选药厂家的检测结果超出相似性验收范围,低于该项质量标准,部分结果见图2,作为较严重的缺陷项可判断为该项不符合相似性判断标准。
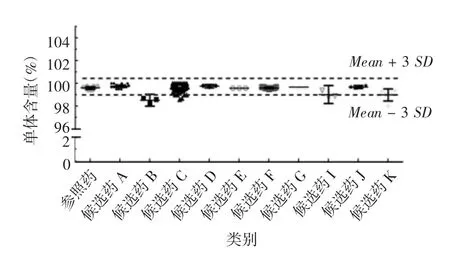
图2 阿达木单抗参照药和候选药的SEC-HPLC纯度
1.4.3.2 CE-SDS纯度 在还原条件及非还原条件下,根据分子量大小的不同采用CE-SDS法进行纯度检测,能检测到降解片段和非糖基化重链,检测结果可反映抗体结构的完整性。各厂家测得阿达木单抗的还原CE-SDS的重链+轻链纯度范围为96.6%~99.3%,非还原CE-SDS的重链+轻链纯度范围为94.1%~100.2%,部分结果见图3。

图3 阿达木单抗参照药和候选药的非还原CE-SDS纯度
1.4.3.3 SDS-PAGE纯度SDS-PAGE可将蛋白质按分子大小分离,包括还原SDS-PAGE和非还原SDSPAGE。各厂家测得阿达木单抗的轻链和重链条带总含量范围为96.6%~99.3%。
1.4.3.4 CEX-HPLC纯度 目前最常采用CEXHPLC检测单抗的电荷异构体,除主峰外的酸性峰和碱性峰对生物活性均有影响。各厂家测得阿达木单抗CEX-HPLC纯度的可见主峰与碱性峰之和均在一定的质量范围内,与文献报道85.62%~87.59%的结果相近[11]。个别候选药的主峰和碱性峰含量超出了初步拟定的相似性标准范围,部分结果见图4,可能与样品前处理有关,建议在头对头对比的基础上尽可能保证电荷异构体与原研药的相似性。对碱性峰含量产生影响的原因之一是C-末端赖氨酸含量,但通常认为由C-末端赖氨酸产生的碱性峰含量的差异不会影响PK及有效性和安全性。

图4 阿达木单抗参照药和候选药的CEX-HPLC纯度
中检院及生物类似药厂家检测参照药的纯度结果与原研自测结果存在一定差异,提示检测方法对结果可能有较大影响,如色谱柱、洗脱条件、样品前处理等,因此生物类似药厂家进行参照药与候选药头对头研究的结果更具有参考意义。
1.4.4 工艺相关杂质 主要包括工艺过程中可能引入的杂质,通常有宿主细胞蛋白质残留量、外源性DNA残留量、蛋白质A残留量及生产过程中其他的添加物(如消泡剂、胰岛素等)。工艺相关杂质并非相似性评价关键项目,主要影响产品的安全性,通常不要求进行头对头对比研究,但应进行工艺相关杂质的去除研究,并对残留量进行安全性风险分析,应不低于已上市同类产品的要求。
2 参照药及候选药的选择标准
2.1 参照药及候选药的选择 药学的质量相似性研究是候选药作为生物类似药开发的前提。应在对参照药的关键质量属性有充分的认识和理解的基础上,采用多种先进、灵敏的技术手段对足够批次的参照药进行全面分析,合理设定质量相似性的验收标准。由于单克隆抗体存在多种翻译后修饰、结构复杂,具有电荷异质性和分子量大小的异质性及批间的不均一性,因此在参照药的选择上,应尽可能收集跨越较长时间范围的多批次来源中国市场的原研药进行表征分析,其他国家来源原研药的质量属性可作为参考,但不作为主要评价依据。候选药应基于质量源于设计的理念,充分考虑生产中使用的原材料、生产工艺、规模、场地、制剂处方、包材等,将具有代表性的批次用于相似性评价的质量研究中[12]。
生物类似药研发中,应纳入尽可能多批次的参照药与候选药,以便相似性分析结果具有统计学意义。申报临床试验阶段,通常应采用至少3批候选药与至少3批参照药进行头对头对比研究;申报上市阶段,应积累10批以上的原研药和候选药的质量研究数据,其中候选药批次中应纳入临床试验批样品、拟上市商业化生产规模批次的代表性样品[13-14]。为确保研发各阶段良好的衔接,关键临床试验前应确定生产工艺、生产规模及生产场地等。
2.2 相似性评价 由于原研药生产工艺信息获取的有限性及生物制品生产工艺的复杂性,难以验证两种来源的生物制品在结构和组成方面的完全一致性,候选药和原研药通常在分子量大小异质性、电荷异质性、糖基化程度方面存在差异。对于质量相似性评价中观察到的差异,应提供更多的支持性资料(包括对安全性、有效性、免疫原性和PK/PD的影响及进一步的研究数据等),来说明存在的差异是否影响候选药与参照药相似性的判断。在某些情况下,可通过部分调整生产工艺来减小或消除观察到的差异,并确保其他的质量属性不会因此受到显著影响[15]。
3 小结
本文基于审评实践所积累的数据,采集了不同生物类药厂家、原研厂家及中检院对阿达木单抗的检测结果。上述数据来源于不同实验室,分析方法、样品来源、样品出厂后放置时间、参照药自身的工艺变更和场地变更等可能存在差异,因此,根据上述数据设定的质量相似性标准范围较为宽泛,虽然对生物类似药厂家的药学研发具有一定参考价值,但建议仅将此标准作为基本参考。对于质量相似性评价,更有意义的是采用灵敏的分析方法对候选药和参照药进行头对头的比较分析。另外,原研药与候选药头对头的图谱比对分析、稳定性变化趋势、降解途径和降解速率的对比也非常重要,但目前采集的数据中,图谱比对和稳定性趋势对比分析的数据有限,本文仅对可用于统计分析的数据进行了梳理,因此得到的质量相似性评价标准具有一定的局限性。生物类似药研发持续深入推进,随着原研药质量研究数据的不断积累及分析技术的进步和统计经验的丰富,生物类似药质量相似性评价标准也会日趋完善。
