以肺动脉高压为首发表现的先天性中枢性低通气综合征临床特征和基因分析
2021-03-29郭予雄吴家兴谭玉玉郑贵浪孙跃玉李渝芬
胡 燕 王 春 郭予雄 王 静 吴家兴 谭玉玉 郑贵浪 孙跃玉 潘 微 李渝芬
广东省人民医院 广东省医学科学院 1.儿科重症监护病房;2.广东省心血管病研究所心儿科(广东广州 510080)
先天性中枢性低通气综合征(congenital central hypoventilation syndrome,CCHS)是类似配对同源基因(PHOX2B)变异导致呼吸中枢化学感受器的敏感性降低,引起肺泡通气不足、自主神经功能失调为主要特征的一种严重而罕见的常染色体显性遗传性疾病[1],多起病于婴幼儿期。其通气不足主要发生在睡眠期间,以非快速眼动睡眠期间最为明显,导致严重的肺泡通气不足,引起高碳酸血症和/或低氧血症。PHOX2B基因变异是CCHS诊断的必要条件[2]。本文报告1例首发表现为重度肺动脉高压的CCHS患儿的临床资料,并结合国内外文献对CCHS 的临床特点、致病机制及基因突变位点进行总结分析。
1 临床资料
患儿,女,11 月龄,因眼睑浮肿2 个月,嗜睡、发绀1周收入广东省人民医院儿科重症监护病房。患儿于入院前2个月无明显诱因下出现眼睑及颜面轻度浮肿,晨起为著,午后消退。入院前1周嗜睡伴发绀、尿少,收治于当地儿童医院,给予气管插管机械通气。期间出现无热抽搐1次,腰椎穿刺测压270 mmH2O;超声心动图提示肺动脉高压(中-重度)。给予西地那非降肺压、利尿、营养心肌及抗感染等对症支持治疗后好转。但多次尝试下调呼吸机参数,患儿均出现发绀、烦躁不安、大汗淋漓,血气分析提示高碳酸血症。为进一步诊治转入我院。患儿系G2P 1,足月顺产,出生体质量3 400 g,无产时窒息史,精神、运动发育与同龄儿相仿。既往健康,无外伤史。父母体健,无家族性遗传性疾病史。入院体格检查:体温36.5 ℃,脉搏94次/min,血压96/68 mmHg,经皮血氧饱和度96%;呼吸机辅助通气(BiPAP模式),吸气峰压(PIP)9 cmH2O,呼吸末正压(PEEP)5 cmH2O,吸入氧浓度(FiO2)30%,吸气时间(Ti)0.75 s,呼吸频率(RR)30次/min。体质量8 kg,头围46.5 cm,发育中等,面容无特殊。前囟张力稍增高,双侧肺呼吸音粗糙,可闻及少许干啰音,心律齐,肺动脉第二音增强,未闻及心脏杂音。腹平软,肝脏肋下2.5 cm,质地软,边缘钝,脾脏肋下未触及,肠鸣音正常。四肢肌力、肌张力正常。生理反射存在,病理反射未引出。实验室检查:血常规无异常;C 反应蛋白、降钙素原、IL-6 均无异常;肺泡灌洗液高通量病原体测序示链球菌序列数高;脑脊液常规、生化检查无异常;血管炎指标无异常;抗核抗体谱正常;血、尿串联质谱分析无异常;丙氨酸氨基转移酶437 U/L;凝血酶原时间22.9 s;B型脑利钠肽25 290.0 pg/mL。彩色超声心动图示三尖瓣反流面积1.5 cm2,估测右室收缩压65 mmHg,肺动脉高压(中-重度),三尖瓣反流(轻度),卵圆孔未闭(图1)。胸部X 线提示右上肺渗出,心影增大,心胸比0.67。心脏CT平扫+增强提示可疑房间隔缺损,肺动脉高压(图2)。腹部B超提示肝脏弥漫性增大,腹腔积液(少量)。头颅MR平扫+增强提示右侧额叶出血(图3)。
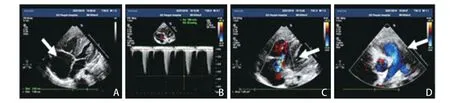
图1 入院第1 天彩色超声心动图表现
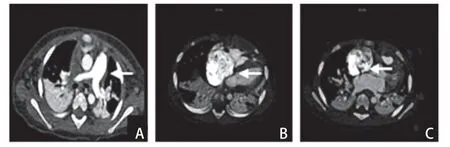
图2 心脏CT 平扫+增强表现
入院后给予镇静镇痛,机械通气,西地那非、安立生坦、瑞莫杜林降肺压,阿司匹林抗凝,强心、利尿及抗感染等治疗。入院第7天彩色超声心动图示右心扩大,三尖瓣局限返流,心脏功能正常。第3周撤机前1小时血气分析示pH值 7.44,PaCO240 mmHg,PaO2155 mmHg,Lac 0.6 mmol/L,予尝试拔管撤机改低流量吸氧,1小时后pH 7.15,PaCO286 mmHg,PaO2240 mmHg,Lac 0.2 mmol/L,遂予无创通气。期间患儿觉醒时自主呼吸规律,睡眠时呼吸不规则、逐渐浅慢、微弱,血气分析示低氧血症及严重二氧化碳潴留,超声心动图示三尖瓣返流(轻-中度)、肺动脉高压(中度)、右室舒张功能稍受限,予上调通气压力支持或呼吸频率恢复正常。入院第4周(出院1个月)复查超声心动图提示三尖瓣反流(轻度),肺动脉高压(轻度)。
患儿临床高度怀疑CCHS,经医院医学伦理审核,并征得父母知情同意后行PHOX2B基因突变分析。抽取患儿及其父母外周静脉血各3 mL 制备基因组DNA,将提取合格的人类基因组DNA 进行文库制备,使用Illumina Hiseq测序平台进行测序分析,测序结果由生物信息学方法进行数据分析。由于PHOX2B基因第3 号外显子GCN 重复区域无法用二代测序方法检测到GCN 的重复数目,所以此区域的拷贝数是否存在异常予行Sanger 法验证。分别对患儿及其父母的PHOX2B基因第3 号外显子GCN 重复区域进行检测,根据PHOX2B基因第3 号外显子GCN 重复序列区域的特点,应用PCR 技术原理进行检测,将扩增产物通过Sanger 测序得到基因片段序列,对该区域GCN 重复数目进行进一步分析,从而判断该区域的具体拷贝数。靶向捕获二代测序未发现可能的致病基因。Sanger 法验证患儿为PHOX2B第3 外显子存在多聚丙氨酸重复扩展突变,基因型为20/25,确诊为CCHS,其父母未检出突变,此变异为新发突变(图4)。
出院后患儿电话随访至26月龄。目前仅夜间需低水平无创BiPAP 正压通气,2 次多导睡眠监测氧合可基本维持稳定,超声心动图示轻度肺动脉高压,生长发育无异常。出院后2 个月头颅MR 平扫+增强提示右侧额叶出血已吸收,局部含铁血黄素沉积。
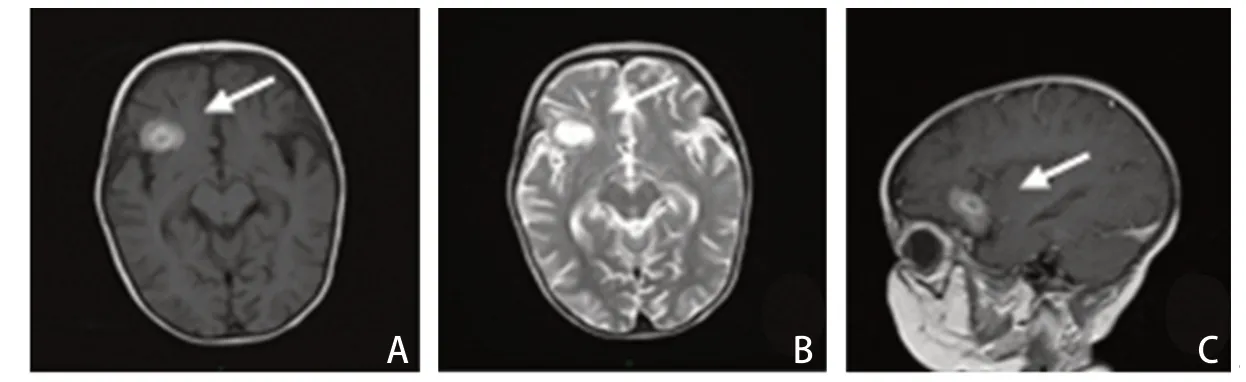
图3 患儿头颅MRI 平扫+增强扫描结果
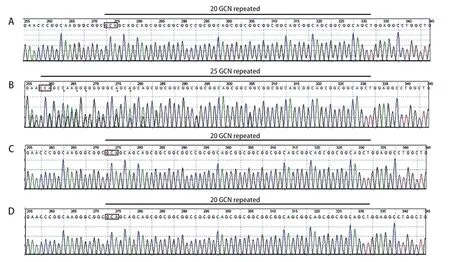
图4 患儿及双亲PHOX2B 基因Sanger 法验证分析
2 讨论
1970 年首次报道具有特征性的儿童CCHS[3],并指出受影响的个体有弥漫性自主神经系统失调(autonomic nervous system dysregulation,ANSD)。CCHS 在一般人群中的发病率尚不清楚,可能存在种族差异。法国发病率约为1/200000[4],日本约为1/148000[5]。国内相关报道甚少,且多为临床诊断病例,2004年报道国内首例儿童CCHS[6]。
CCHS 被归入呼吸系统疾病,实际上是一种罕见的神经系统疾病,与脑干神经嵴畸形有关,由于迷走神经传入丧失和髓质CO2受体敏感性降低导致睡眠期间呼吸的自主控制受损,睡眠时通气不足。患儿对高碳酸血症和低氧血症敏感性降低导致睡眠相关性低通气,表现为慢性呼吸功能不全[1,7-8],常可伴先天性巨结肠、神经嵴源性肿瘤和自主神经系统功能障碍。CCHS 患者在睡眠期间表现出最严重的呼吸调节失调,并且对高二氧化碳或低氧没有表现出正常的生理反应,不出现呼吸窘迫或其他缺氧反应,结果血气紊乱持续进展,至清晨清醒前最为严重[9-10],这也为CCHS 早期诊断增加了难度。因低通气的严重程度各不相同,CCHS可发生在任何年龄,90%典型CCHS于新生儿期起病,伴有呼吸暂停、发绀或高碳酸血症,大部分需机械通气,但反复拔管失败。较轻的病例可于儿童或成年后才被诊断(晚发CCHS)[1,9,11-12]。患者可能由于未察觉的低氧血症和/或高碳酸血症而并发癫痫发作、红细胞增多症,部分可出现多汗、水肿和右心衰竭,易被误诊为先天性心脏病等其他疾病。对于大龄儿童和成人,肺动脉高压和肺心病可能是CCHS的先兆[13]。据报道,CCHS 中有78%的肺心病发生率[14]。本例患儿以重度肺动脉高压为首发表现,国内外鲜有报道。关于CCHS并发肺动脉高压的机制尚不清楚,可能因患儿长期肺泡低通气、缺氧造成肺血管痉挛,严重者可发生肺动脉高压、右心功能不全。目前尚无有效药物根治该病。早期识别和通气干预可以逆转慢性缺氧诱导的心力衰竭[15]。
2010年美国胸科学会发布CCHS基因学、诊断和管理共识[1],提出基因检测在CCHS 诊断和治疗中的重要作用,其可预测疾病严重程度分型及可能的并发症。对于原因不明的持续高碳酸血症和低氧血症的儿童应高度警惕此病,采用多导睡眠仪有助于早期发现呼吸异常。2003 年发现PHOX2B是CCHS 的致病基因[12],有研究者指出PHOX2B为CCHS 疾病定义的基因,PHOX2B基因变异是诊断CCHS的必要条件[16]。该基因位于染色体4p12,编码1个高度保守的同源盒结构域转录因子,具有2条聚丙氨酸链,由9和20个聚丙氨酸和1个同源异型盒组成。PHOX2B基因对呼吸中枢的发育、自主神经系统的分化和诱导起着至关重要的作用[16-17]。CCHS患儿中枢神经系统调节呼吸通路存在缺陷,而该通路的发生和运作受PHOX2B基因调控。PHOX2B序列变异的检测对于确诊CCHS有重要意义,尤其对于症状不典型的患儿。
超过90%的CCHS 患者为PHOX2 B聚丙氨酸重复扩展变异(polyalanine repeat expansion mutation,PARM)杂合子,范围从24到33个丙氨酸,产生20/24至20/33 基因型(正常基因型为20/20),其中基因型20/25、20/26和20/27最常见[1]。其余10%的患者为杂合变异(no-polyalanine expansion mutation,NPARM),包括错义、无义和移码变异[18]。携带PARM 的患者临床严重程度随丙氨酸重复序列的增加而增加[5,19]。PARM 的基因型与持续通气依赖有关,随着丙氨酸重复序列的增加,需24小时辅助通气的可能性越大。而大多数NPARM 患者在新生儿期就表现出低通气症状,并伴有先天性巨结肠、自主神经系统调节失调和神经嵴起源肿瘤,故对此类患儿应仔细检查各种并发症[20-21]。关于基因型-表型关联的数据可能有助于疾病的临床管理。了解患者的确切突变可以对CCHS 并发症进行准确预测和针对性筛查[22]。
本例患儿为PARM,基因型为20/25,为常见基因型,发病较晚,仅夜间需低水平无创通气。因携带基因型20/25 的成年人可能出现不完全和可变的外显性,以及轻微到中度的临床症状,建议任何具有基因型20/25 CCHS 先证者的父母都进行PHOX2B检测[13]。本例患儿父母PHOX2B基因测序虽未检测到相同变异和体细胞嵌合体,患儿的变异考虑为新发生的变异,但仍然不能排除父母存在生殖腺嵌合体的可能。建议患儿父母再次生育时需进行产前基因诊断[23]。
CCHS目前尚缺乏有效改善疾病相关低通气的药物,患者终生需通气治疗。未经治疗者因长期肺泡低通气、严重缺氧可致肺动脉高压、右心功能不全。对于不明原因的肺动脉高压患儿需观察呼吸情况;疑诊者在除外可导致低通气的心、肺、神经肌肉功能障碍原发病后,如常规靶向捕获二代测序未发现可能的致病基因,应尽早行PHOX2B基因Sanger法验证分析,可避免漏诊CCHS,并预测疾病严重程度分型及可能的并发症。早期给予无创通气可改善预后。
