新生儿瓜氨酸血症I 型一家系基因分析
2021-03-29陈雨晗张万巧
彭 薇 杨 晓 陈雨晗 闫 磊 张万巧
中国人民解放军总医院第七医学中心 出生缺陷防控关键技术国家工程实验室 儿童器官功能衰竭北京市重点实验室(北京 100700)
瓜氨酸血症是一种常染色体隐性遗传代谢病,包括瓜氨酸血症Ⅰ型(citrullinemia type Ⅰ,CTLN1)和瓜氨酸血症Ⅱ型(CTLN 2)两种类型,CTLN 1 为尿素循环障碍疾病之一,由精氨基琥珀酸合成酶(argininosuccinate synthase,ASS1)缺乏所致的先天性遗传代谢性疾病,以瓜氨酸血症及高氨血症为主要特征。CTLN1全球新生儿中的发生率约为1/250 000[1],临床以急性高氨血症,血浆瓜氨酸升高,精氨酸降低为主要特征。根据临床特征,CTLN1又可分为早发型和迟发型两大类,早发型在生后几天就出现明显临床症状,常表现为嗜睡、喂养困难、呕吐、癫痫和意识丧失,预后差,死亡率高;迟发型一般在青春期或成年期发病,比较少见,个别轻型迟发型可终生无症状。目前国内仅报道11例CTLN1的ASS1基因检测结果,发现了16个变异位点,在新生儿期报道的则更为稀少。本研究对1例新生儿CTLN1家系进行基因分析,探讨其发病的遗传学病因,丰富中国人新生儿ASS 1基因变异谱,并为遗传咨询提供依据。
1 临床资料
患儿,女,生后6天,G1P1,39+2周顺产,出生体质量2 600 g,Apgar评分10分。父母非近亲结婚,表型无异常,家族史、母亲妊娠史均无异常。患儿因出生后纳差、反应差、反复哭闹入院,入院后出现抽搐;常规生化检查示血氨升高、血乳酸升高、血糖降低。患儿病情持续加重,逐渐出现呼吸衰竭、心肌损伤、肝肾功能异常。血串联质谱检查提示瓜氨酸显著增高,1 621 μmol/L(正常参考值2.79~15 μmol/L)。尿液气相色谱质谱检查提示尿乳清酸显著增高,响应值为4.54(正常上限响应值0.04),尿嘧啶浓度增高,响应值为1.10(正常上限响应值0.08)。参考本实验室代谢生化平台建立的诊断标准,定义响应值为检测化合物的峰面积与内源性肌酐峰面积的比值,正常上限值为代谢正常新生儿各化合物响应值的第99.5百分位数[2]。结合患儿临床表现,以及血尿质谱检测结果,提示患儿为CTLN1。
为进一步明确诊断及进行遗传咨询,经解放军第七医学中心伦理委员会批准,患儿父母知情同意后,采集患儿及其父母EDTA 抗凝外周血进行基因诊断。应用北京Tiangen公司的TIANamp Blood血液基因组DNA提取试剂盒(目录号:DP318)提取基因组DNA,使用标准文库构建试剂盒(北京迈基诺基因科技股份有限公司)进行基因组文库构建。质检合格后采用目标序列捕获探针(北京迈基诺基因科技股份有限公司)对包含ASS 1基因在内的175 个已知代谢疾病候选基因外显子区域进行捕获,获得目标基因的富集文库。借助Illumina NextSeq 500测序平台对富集文库进行高通量测序,获得原始测序数据,通过生物信息分析和变异位点的筛选出具报告。结果发现患儿ASS 1基因发生c.1194-2A>G杂合变异和3号外显子杂合缺失。家系验证发现患儿父亲ASS1基因发生c.1194-2A>G杂合变异,患儿母亲ASS1基因3号外显子杂合缺失。对此剪切变异进行查询,在HGMD、ESP6500、千人基因组(1000g2015aug_ALL)和ExAC等数据库均未见报道,该变异不属于多态性位点,在人群中发生频率极低,根据ACMG指南划分为可能致病性变异(PVS1+PM2)。
为验证二代测序结果,首先对剪切突变进行Sanger测序,引物合成由上海生物工程有限公司完成,PCR反应体系扩增产物进行Sanger测序分析,结果发现,患儿及其父亲ASS1基因发生c.1194-2A>G杂合变异,患儿母亲ASS1基因测序未检测到变异,见图1。为进一步验证剪切变异对蛋白功能的影响,通过HSF、MutationTaster和regSNP-intron三个软件对剪切变异进行生物信息学分析,预测结果分别为破坏变异位点、引发疾病、可能引发疾病,推测此变异为可能致病性变异。最后对外显子3 进行定量PCR 试验,引物序列分别为,exon3-F:GAA GGA ACA AGG CTA TGA CG,exon 3-R:CAG GAC ACG CAG GAA GAC;exon4-F:CCT CTC CGC TTC TGC TTC,exon4-R:CCA AGC TTC AGT GCC TTC。将Roche480的结果输出到Excel进行分析,利用Excel中的公式“=POWER(2,-ΔΔCt)”得到最终结果,见图2。定量PCR 结果也显示患儿及其母亲ASS1基因3号外显子杂合缺失,进而说明3号外显子杂合缺失是患儿另外一个可能性致病性变异。
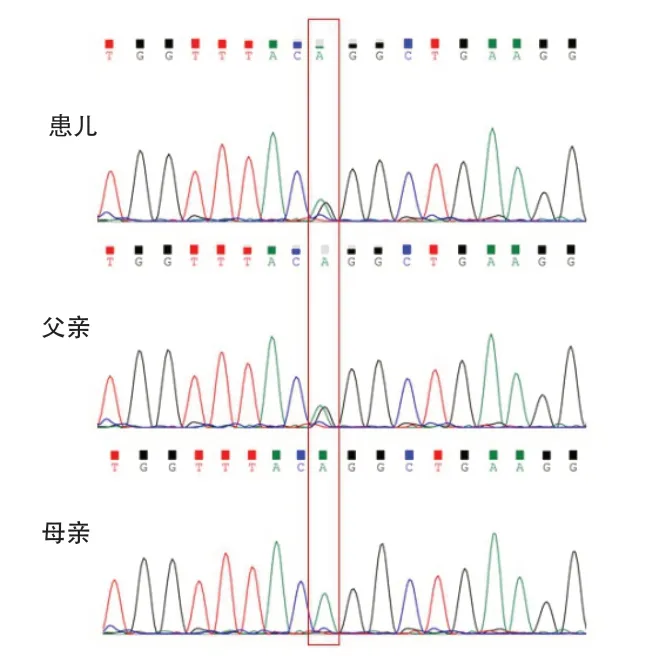
图1 ASS1 基因Sanger 测序图
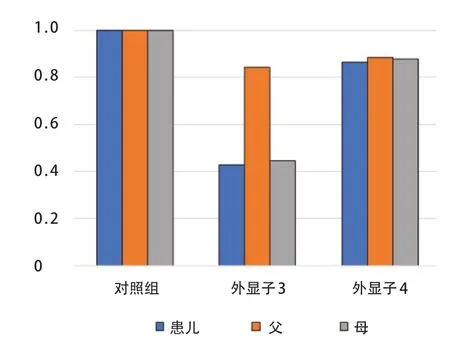
图2 外显子3、4 定量PCR 统计图
2 讨论
CTLN 1 是常染色体隐性遗传病,发病率位居尿素循环障碍中的第3 位。临床表现轻重不一,重症患儿多在新生儿期发病,多在进食蛋白质饮食后逐渐出现喂养困难、精神萎靡、嗜睡、昏睡,甚至惊厥昏迷,血液中瓜氨酸及氨含量明显升高[3]。本例患儿出生无异常,起病急、病程短,出现难以纠正的高血氨和高乳酸,高度怀疑遗传代谢病。经完善血串联质谱及尿气相质谱检测,符合CTLN 1 诊断。CTLN 1 由ASS 1基因变异引起。ASS1基因位于染色体9q34.1,长度约为63kb,具有1259 bp开放阅读框,包含16个外显子,起始密码子位于外显子3中,终止密码子位于外显子16中,共编码412个氨基酸[4],催化瓜氨酸及天冬氨酸生成精氨酰琥珀酸的精氨酰琥珀酸合成酶,是尿素循环中重要的限速步骤。CTLN 1 确诊主要依据ASS 1基因或ASS 酶活性检测。ASS 酶肝脏活检为侵入性操作,对新生儿伤害较大,其他间接检测方法准确性低[5];而基因检测不仅可以明确先证者的临床诊断,还对之后可能进行的产前诊断至关重要。
本研究利用二代测序发现患儿ASS 1基因存在c.1194-2 A>G 和3 号外显子缺失的复合杂合变异。家系验证显示,c.1194-2A>G变异来源于父亲,3号外显子缺失来源于母亲,在HGMD、ESP 6500、千人基因组、EXAc 等数据库中均未见报道。利用HSF、MutationTaster和regSNP-intron软件对c.1194-2A>G变异进行生物信息学分析,预测结果分别为破坏变异位点、引发疾病、可能引发疾病,说明该剪切变异可能对蛋白功能有影响,但仍需进一步功能学研究来验证。
截止目前,HGMD 数据库共报道ASS 1基因变异163种,其中错义变异最为常见,大约114种,剪切变异16种,大片段缺失变异10种。剪切变异分布在除1、3、8、9、10、14之外的其余内含子上,第15内含子上报道过1种剪切变异。早在1990年就发现ASS1基因第15内含子上发生c.1194-1G>C剪切变异[6],与本例患儿的变异仅相差一个碱基,该研究通过cDNA分析推测变异会导致3种异常剪切事件发生,变异导致精氨酸琥珀酸酶C-端读码框发生移位,影响蛋白质的最后15个氨基酸,从而导致酶活性丧失,充分证实C-端序列对于维持蛋白质稳定性至关重要。2004年又发现1例孕妇携带c.1194-1G>C剪切变异[7]。
HGMD数据库报道的ASS1基因发生大片段缺失的仅10种,在新生儿期检测到的外显子缺失分布在5、6、7、8、9、11、12和13外显子上。早在20世纪,美国贝勒医学院和日本鹿儿岛大学就先后在新生儿瓜氨酸血症患者中检测到外显子5、6、7和13缺失[5,8-9];2008年报道1例波兰新生儿外显子11和12缺失[10];2019年在141例新生儿干血片筛查阳性患儿基因诊断中发现1例大片段杂合缺失变异,缺失范围包含外显子8和9[11]。国内相关研究报道非常有限,已报道新生儿患者仅5例,共发现7种变异位点,变异类型包括纯合变异和复合杂合变异(两个不同的错义变异、错义变异和剪切变异、错义变异和移码变异),患儿均在出生后2~6天发病,临床结局差[1,12-16]。结合本例患儿及已有报道,中国CTLN 1 新生儿患者变异谱具有异质性,未发现高频或热点突变。大多数中国患儿属于复合杂合状态,基因型和表型间的关联研究困难。推测本例患儿的2种新发变异与急性血氨升高和预后较差相关。
本研究显示,二代测序可同时对于剪切变异和大片段的缺失进行准确分析,Sanger测序对点突变有较好的验证效果,定量PCR对杂合缺失有较好的检测效果,生物信息学分析能较好地预测剪切变异对蛋白功能的影响,多技术的联合运用可以提高ASS 1基因变异的检出率,也为产前诊断和遗传咨询提供了遗传依据。同时进行代谢分析和基因检测不仅可以相互佐证结果的准确性,而且可以提高新生儿遗传代谢病的确诊率。
综上,本研究分析1例CTLN1新生儿的临床表型、代谢表型及基因型,发现两种新发变异,丰富了中国新生儿ASS1基因变异谱。对早发型CTLN1应尽早完善血串联质谱、尿液气相质谱检查,必要时联合多种基因检测技术进行基因分析以明确诊断,早期救治,提高患儿的生存率,同时为进一步的产前诊断和遗传咨询提供可靠依据。
