EARS2 基因变异致新生儿丘脑脑干受累性脑白质病伴高乳酸血症1 例报告并文献复习
2021-03-29崔清洋唐成和桑桂梅曹银利王卫卫刘育红
崔清洋 唐成和 桑桂梅 曹银利 王卫卫 刘育红
1.新乡医学院第一附属医院儿科(河南卫辉 453100);2.河南省淇县人民医院儿科(河南淇县 456750)
线粒体疾病是一组遗传和临床异质性疾病,通常具有进行性和多系统性受累的特点,为人类最常见的遗传性疾病之一。细胞基质中的线粒体受核基因组及线粒体基因组的双重遗传调控,其功能是通过氧化磷酸化给正常细胞提供所需能量。已有越来越多的证据显示,氧化磷酸化系统缺陷是任何器官或系统各种临床表现的原因,从产前并发症到急性新生儿失代偿和死亡到成人发病的疾病[1]。有报道证实,许多诊断为氧化磷酸化障碍的产前异常患儿病因有宫内生长迟缓和各种胎儿畸形[2]。一些“新生儿综合征”可能由氧化磷酸化障碍所致,如致命的新生儿线粒体疾病[3]。在这些综合征中,如肝线粒体DNA 缺失引起的严重肝病,其临床表现与酶或分子缺陷之间的关系已被证实。氧化磷酸化障碍有关的新生儿表现特征也可能在新生儿期之后出现,例如辅酶Q10缺乏症或涉及肝功能不全的疾病。
丘脑脑干受累性脑白质病伴高乳酸血症(leukoencephalopathy with thalamus and brainstem involvement and high lactate,LTBL)是由EARS2基因变异导致的常染色体隐性遗传线粒体脑病。LTBL 临床表型差异很大,有发育迟缓;眼部受累有眼睑下垂、眼外肌麻痹、视觉障碍;口唇受累可有腭裂;心脏受累可有室间隔肥厚;肝脏受累有肝大、脂肪变性、纤维化及胆汁淤积;肌肉及软组织受累可有新生儿肌张力低下,肌组织活检见破碎红肌纤维及COX C氧化酶阴性肌纤维,尚有线粒体复合体Ⅰ、Ⅱ、及Ⅳ活性下降;中枢神经系统受累有精神运动退化(较轻病例),痉挛性四肢瘫、肌张力障碍、运动迟缓、头部控制较差、精神运动发育迟缓(均见于严重病例)及癫痫发作;代谢异常有高乳酸血症;头颅MRI 可见深部白质肿胀,脑深部白质、脑干及伴脑室周围完整的小脑白质T 2 加权高信号,胼胝体变薄及胼胝体发育不良。实验室检查偶有间歇性肝酶异常及甲胎蛋白升高。
本文回顾分析1 例LTBL 新生儿的临床资料及基因检测结果,并首次报道EARS 2核基因c.1294 C>T无义变异及c.971G>T错义变异,以期加强对新生儿线粒体疾病的认识。
1 临床资料
患儿,女,出生26 小时,因反应差伴呼吸呻吟5小时入院。患儿出生20 小时余出现不明原因的反应差、呼吸呻吟,伴奶量少及睡眠增多,并在5小时内发生双眼凝视及颜面发绀2 次,2~3 s/次,无发热及呕吐,无尖叫,血糖1.7 mmol/L。以新生儿低血糖症、先天性心脏病、新生儿呼吸窘迫综合征收住院。血气分析示有危及生命的代谢性酸中毒及高乳酸血症,予经鼻持续呼吸末正压通气、纠正低血糖、纠酸及静脉营养治疗,低血糖纠正,但呼吸困难及酸中毒逐渐加重转诊至新乡医学院第一附属医院。患儿系G2P 1,足月顺产,产前胎膜早破6 小时,羊水Ⅱ度污染,出生时无产伤及窒息史。患儿父母体健,非近亲结婚,母亲第1 胎于妊娠6 个月时因胼胝体缺如人工流产,流产胎儿组织行染色体微阵列检测未见异常。入院体格检查:一般情况极差,神志不清,反应极差,面色青灰,全身皮肤发灰,前囟平坦,口周发绀,气管插管状态,自主呼吸弱,双肺呼吸音粗,心前区可闻及5/6收缩期杂音,腹部饱满,肝脾肋下未触及,肠鸣音弱,四肢肌张力低下,原始反射未引出。产前4 个月,外院胎儿头颅磁共振成像(MRI)示胼胝体缺如;产前2 个月本院胎儿头颅MRI 示胼胝体缺如。外院血气分析示pH 值6.796,氧分压54.6 mmHg,二氧化碳分压39.2 mmHg,钾4.7 mmol/L,钠147 mmol/L,氯113 mmol/L,钙1.34 mmol/L,乳酸20.0 mmol/L,碳酸氢盐6.0 mmol/L,剩余碱28.0 mmol/L。入院后纠酸期间(血液净化前)血气分析pH 值波动于6.76~7.16,乳酸波动于15.1~31.2 mmo/L,碳酸氢盐波动于4.5~7.8 mmo/L,剩余碱波动于19.6~28.5 mmo/L;血液净化期间pH 值波动于6.97~7.58,乳酸波动于9.0~28.5 mmo/L,碳酸氢盐波动于5.4~18.9 mmo/L,剩余碱波动于28.1~8.4 mmo/L;血液净化后血气分析pH 值7.13,氧分压162 mmHg,二氧化碳分压28.4 mmHg,钾3.88 mmol/L,钠149 mmol/L,氯105 mmol/L,钙1.13 mmol/L,乳酸30.9 mmol/L,碳酸氢盐9.1 mmol/L,剩余碱18.7 mmol/L。血常规白细胞22.5×109/L,血红蛋白150 g/L,血小板258×109/L;超敏C反应蛋白11.80 mg/L;降钙素原0.86 ng/mL;脑钠肽前体28 400 pg/mL;总蛋白(TP)54.4 g/L,白蛋白(ALB)35.4 g/L,总胆红素(TBIL)93.2 mol/L,间接胆红素(IBIL)93 umol/L,丙氨酸氨基转移酶(ALT)30U/L,天冬氨酸氨基转移酶(AST)267U/L,谷氨酰转肽酶(GGT)129U/L;尿素2.02 mmol/L,肌酐91.8 mol/L,尿酸672μmol/L;乳酸脱氢酶(LDH)4 320 U/L,肌酸激酶(CK)6 850 U/L,肌酸激酶同工酶(CKMB)580 U/L。胸片示两肺透过度稍减低、纵隔增宽,心影稍大。彩色多普勒超声心动图示二尖瓣(中度)、三尖瓣(重度)关闭不全、肺动脉高压(60 mmHg)、卵圆孔未闭。入院诊断:代谢性酸中毒,高乳酸血症,脓毒血症,肺动脉高压,新生儿吸入性肺炎,高磷血症,心肌损害,遗传代谢性疾病,胼胝体缺如。
入院后予患儿呼吸机辅助通气、静脉营养、营养心肌及脑细胞、抗感染、纠正酸中毒治疗。入院1 天后复查电解质,血钾4.08mmol/L,钠142 mmol/L,氯86.6 mmol/L,钙1.77 mmol/L,二氧化碳5.0 mmol/L,磷3.90 mmol/L,镁1.19 mmo/L;TP 49.1g/L,ALB 32.4g/L,TBIL 81.0 µmol/L,IBIL 76.3 µmol/L,ALT 48 U/L,AST 470 U/L,GGT 74 U/L;尿素3.45 mmol/L,肌酐162.2 µmol/L,尿酸868 µmol/L;LDH 7 510 U/L,CK 11 460 U/L,CKMB 1 460 U/L;因乳酸持续升高及急性肾损伤,给予11小时床旁血液净化治疗后乳酸下降不明显。因病情进行性加重,住院2 天后家属放弃治疗后死亡。
因患儿持续高乳酸血症,高度怀疑遗传代谢性疾病,行血串联质谱及尿气相-色谱质谱检测,示多种氨基酸及酰基肉碱增高,伴乳酸、2-羟基丁酸、丙酮酸、3-羟基丁酸及3-羟基异丁酸增高。因患儿顽固性高乳酸、意识障碍及抽搐并结合血串联质谱及尿气相-色谱质谱结果高度怀疑线粒体疾病可能。经医学伦理审核及家长知情同意后,在患儿死亡前采集患儿外周血4 mL及其父母外周血2 mL行基因检测。结果显示,患儿线粒体EARS2核基因第7号外显子c.1294C>T杂合无义变异和第5 外显子c.971 G>T 杂合错义变异,组成复合杂合变异(图1、2)。此两种变异可能导致蛋白质功能受到影响,但2个变异的致病性尚未见文献报道(参考数据库:HGMD Pro及PubMed)。家系验证结果显示,c.1294C>T遗传自父亲,c.971G>T遗传自母亲,符合常染色体隐性遗传规律。
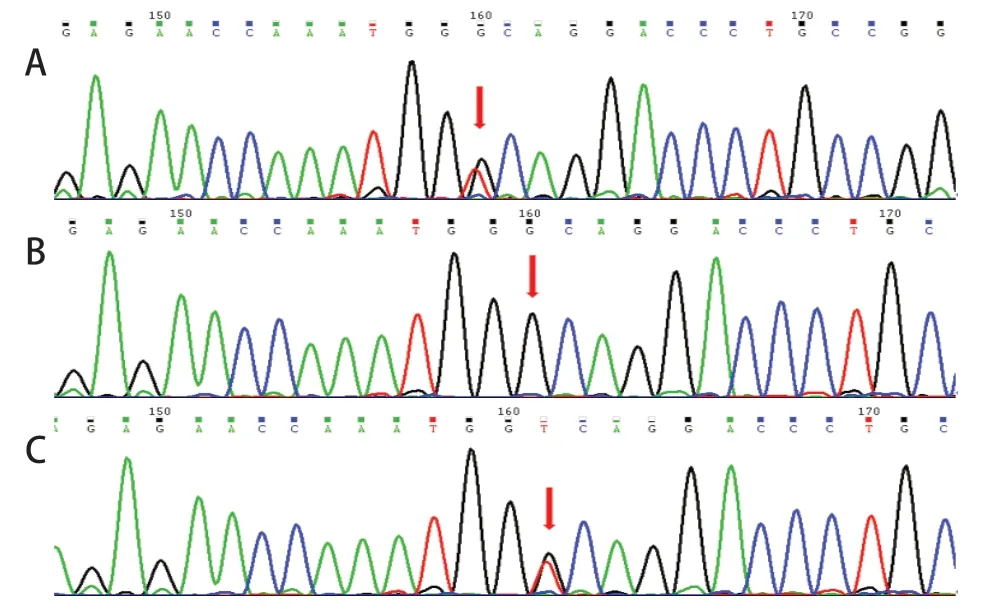
图1 EARS2 基因c.1294C>T 变异测序峰图
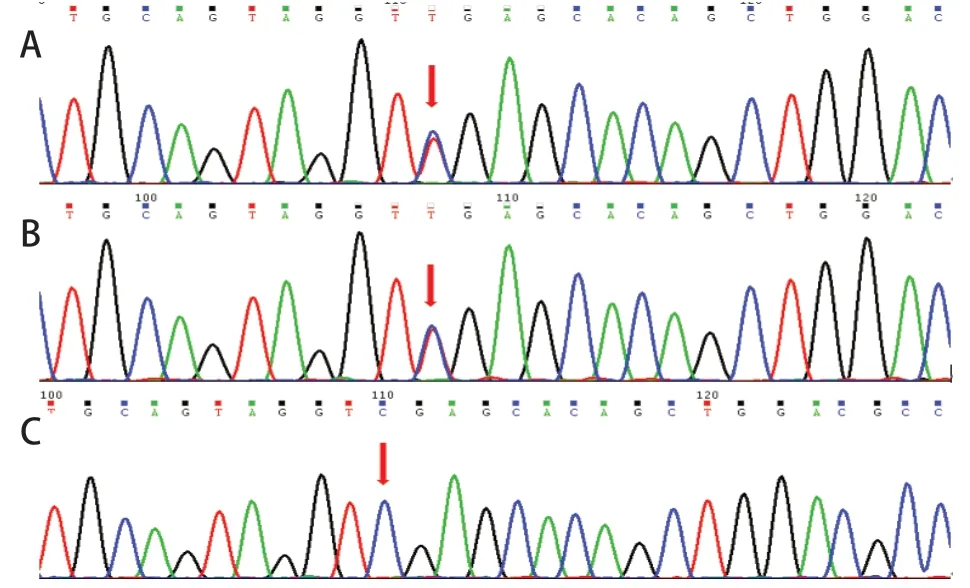
图2 EARS2 基因c.971G>T 变异测序峰图
线粒体基因二代测序未发现患儿有临床意义的线粒体基因变异,但MLPA 检测发现患儿及其母亲线粒体基因MTND1-1*异质性缺失变异(图3)。
根据美国医学遗传学与基因组学学会(ACMG)联合美国分子病理学会(AMP)2015年制订的基因序列变异的解释标准和指南进行致病性分析[5]。EARS2基因c.1294C>T的致病性:①c.1294C>T变异为无义变异,为致病变异(非常强致病性证据,PVS 1);②c.1294C>T变异通过比照千人基因组数据库(1000 Genomes)、人类基因变异数据库(HGMD)未见收录(中等致病性证据,PM 2);③经多种算法预测会对基因或基因产物功能造成有害影响的c.1294 C>T 变异(支持致病证据,PP3);④综合上述c.1294C>T变异的证据强度为“PVS1+PM2+PP3”,判断为导致受检者发病的致病性变异。EARS 2基因c.971 G>T 的致病性:①c.971 G>T 变异位于变异热点和/或位于已知无良性变异的关键功能区域(中等致病性证据,PM1);②c.971G>T变异通过比照千人基因组数据库(1000 Genomes)、人类基因变异数据库(HGMD)未见其收录(中等致病性证据,PM2);③经多种算法预测会对基因或基因产物功能造成有害影响的c.971G>T变异(支持致病证据,PP3);④综合上述c.971G>T变异的证据强度为“PM1+PM2+PP3”,判断为临床意义不明的变异。
综合患儿临床表现及基因检测分析,诊断线粒体EARS 2核基因c.1294 C>T 变异及c.971 G>T 变异所致的LTBL 基本明确,MLPA 检测发现的患儿线粒体基因MTND1-1*异质性的缺失变异加重了病情。
2 讨论
人类线粒体疾病约40%在新生儿期发病,遗传方式中有25%为母系遗传,大部分为常染色体隐性遗传,时有散发、X 连锁和常染色体显性遗传。线粒体内的氧化磷酸化系统包括线粒体的呼吸链复合体(复合体Ⅰ~Ⅳ)和腺苷三磷酸酶(复合体Ⅴ),其中线粒体基因编码13 个亚基,而核基因编码70 余个亚基。
线粒体氨酰基tRNA 合成酶是一组核基因所编码的酶,在遗传信息的翻译中发挥至关重要的作用。到目前为止,已经鉴定出17 种线粒体特异性氨酰基RNA 合成酶,分别为AARS 2、DARS 2、RARS 2、NARS 2、CARS 2、EARS 2、HARS 2、IARS 2、LARS 2、MARS 2、FARS 2、PARS 2、SARS 2、TARS 2、WARS 2、YARS 2和VARS 2基因所编码。而这些基因中的多数(AARS2、DARS2、RARS2、NARS2、CARS2、EARS2、HARS 2、IARS 2、LARS 2、MARS 2、FARS 2、SARS 2、TARS 2、YARS 2和VARS 2)与婴儿发病的线粒体疾病发展有关。
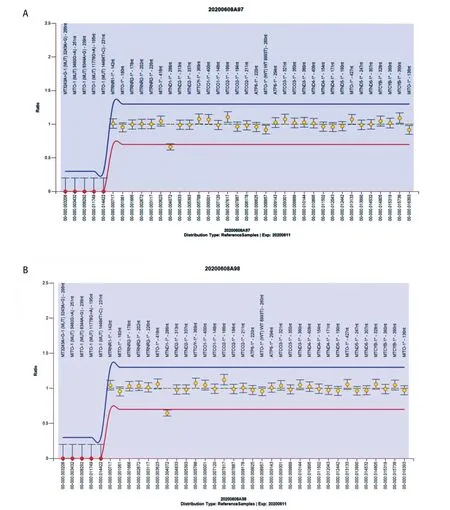
图3 患儿及母亲线粒体MLPA 检查结果
线粒体氨酰基tRNA 合成酶遗传缺陷代表了线粒体疾病的一个新的亚组,可以表现为不同的临床表型。其相关的症状包括脑病、肌病/心肌病、贫血、肾小管病变、听力损失和女性卵巢发育不全。但值得注意的是,该病临床预后往往不良且无具体的治疗选择可用。
然而除了上述的特征外,到目前为止报告的儿童中可确定3个临床亚组[6]:①新生儿期/早期婴儿发病的严重表型,即乳酸性酸中毒、脑病、严重的肌张力低下和包括严重胼胝体发育不全在内的大脑异常[4]。到目前为止报道的6例患儿中2例预后严重不良[7-8],另外有研究报道胼胝体严重累及似乎是预后不良的一个指标[6];②中间表型,在出生的前6个月内发病,表现为与第1种表型类似的临床特征,但临床和神经影像学特征可自发恢复,目前仅有1 例报道[9];③相对温和表型,出生6 个月后发病,表现为乳酸升高和精神运动发育迟缓。值得注意的是,这些患儿神经影像学本身就不太严重,且疾病呈现自发的临床和生化改善及MRI异常的消退[4]。除了以上报道的临床表型外,最近有报道将1例儿童丘脑缺失也描述为EARS2基因变异的放射学特征[10]。
经过文献检索,目前全球共报道27 例(国内1例[11])LTBL,均在出生后1 年内发病,临床特征包括血乳酸升高及包括胼胝体发育不良及脑白质、丘脑、中脑、脑干和小脑白质对称性改变在内的颅脑影像学异常。27例中新生儿期发病6例,死亡4例,1例于出生21天死亡。新生儿期死亡病例为1例男性患儿,出生后不久即出现低血糖(1.6 mmol/L),数小时稳定期后出现严重的乳酸性酸中毒(pH 值6.97,乳酸25.2 mmol/L),予碳酸氢钠治疗后酸中毒纠正。因怀疑遗传性代谢缺陷所致反复低血糖发作,予葡萄糖泵注维持血糖稳定,10 mg·kg-1·min-1,但下调糖速后血糖再次下降。头颅超声显示胼胝体发育不全;血、尿和脑脊液高乳酸、血磷酸肌酸和丙氨酸及脯氨酸升高均指向线粒体疾病的可能。因继发肉碱缺乏予经验性的维生素K1和肉碱补充治疗。此后患儿在持续泵入葡萄糖和碳酸氢钠后乳酸水平有所改善,但从未恢复正常;之后患儿病情恶化,乳酸再次升高到252 mmol/L,最后在21天时死于心肺衰竭。基因检测发现其EARS 2基因c.320 G>A 和c.328 G>A 复合杂合变异。本例患儿出生后不久出现低血糖及顽固性代谢性酸中毒及高乳酸血症,胎儿头颅MRI提示胼胝体缺如,结合基因检测结果,新生儿LTBL诊断明确。
造成LTBL 的EARS 2基因变异的类型较多,有碱基缺失、置换及插入。迄今所报道导致LTBL 的EARS2变异大多为复合杂合变异,仅有5例为纯合变异,但尚未明确基因变异位点与疾病临床的严重程度两者关系,然而纯合变异的临床表型更为严重。研究报道,EARS2基因纯合变异的病情通常进展快速并死亡,可能和线粒体呼吸链复合体Ⅰ及Ⅳ的严重缺乏关联[7]。本例患儿为EARS2基因c.1294C>T和c.971G>T复合杂合变异,支持文献报道。
综上,报道1例首次发现的EARS2基因c.1294C>T和c.971 G>T 复合杂合变异致LTBL 患儿,扩充了LTBL 的基因变异谱。但如需进一步明确EARS 2基因c.971 G>T 变异位点的致病性,需要进行大样本的回归分析并行转基因动物的模型验证。
