奥卡西平刻痕片仿制药与原研药溶出行为及相关指标的一致性评价研究
2020-07-01吕蓓蓓杨海源程华魏文芝张敏娟
吕蓓蓓 杨海源 程华 魏文芝 张敏娟


中圖分类号 R917 文献标志码 A 文章编号 1001-0408(2020)12-1463-08
DOI 10.6039/j.issn.1001-0408.2020.12.11
摘 要 目的:评价奥卡西平刻痕片仿制药与原研药溶出行为的一致性,并比较两者的外观、半片制剂的脆碎度及分割质量损失以及不同企业原料药的晶型、晶体形貌。方法:采用高效液相色谱法测定含量;采用桨法(设置转速60 r/min,温度37.0 ℃)测定仿制药与原研药在4种不同溶出介质[含0.6%十二烷基硫酸钠的盐酸溶液(pH=1.2)、含0.6%十二烷基硫酸钠的醋酸盐缓冲溶液(pH=4.5)、含0.6%十二烷基硫酸钠的磷酸盐缓冲溶液(pH=6.8)、含0.6%十二烷基硫酸钠的水溶液]中的累积溶出度,采用相似因子法评价两者溶出曲线的相似性,并评价半片与整片制剂的批内均一性;采用脆碎度检测仪及电子天平测定半片制剂的脆碎度及分割质量损失;采用X射线衍射仪及扫描电子显微镜观察不同企业原料药的晶型和晶体形貌。结果:奥卡西平检测质量浓度的线性范围为33.42~401.09 μg/mL(r=0.999 9);定量限为0.10 μg/mL,检测限为0.04 μg/mL;精密度、稳定性、重复性、耐用性试验的RSD均小于2%;回收率为99.80%~101.63%(RSD为0.37%~0.91%,n=3)。仿制药A、B和原研药在4种不同溶出介质中90 min时的平均累积溶出度分别为92%、87%、90%[含0.6%十二烷基硫酸钠的盐酸溶液(pH=1.2)];94%、94%、90%[含0.6%十二烷基硫酸钠的醋酸盐缓冲溶液(pH=4.5)];95%、95%、91%[含0.6%十二烷基硫酸钠的磷酸盐缓冲溶液(pH=6.8)];97%、98%、95%(含0.6%十二烷基硫酸钠的水溶液)。仿制药A、B和原研药在上述4种溶出介质中的相似因子分别为66、81,71、69,71、61,59、39。前15 min时,仿制药A、B和原研药半片与整片的溶出度差值分别为-3%~13%、-2%~24%、-3%~7%;仿制药A半片、整片累积溶出度的RSD分别为6%~14%、2%~9%(n=12),仿制药B为4%~10%、1%~8%(n=12),原研药为2%~7%、2%~8%(n=12)。原研药外观为梭形,刻痕较深;仿制药片形各异,且刻痕明显浅于原研药。仿制药A、B和原研药的脆碎度、分割质量损失分别为0.62%、0.67%,0.12%、0.11%,0.08%、0.05%。国产原料药呈不规则的块状和碎屑,原研药企业产原料药呈规则的扁平长方体和规则的条状且碎屑少;但两者的X射线衍射特征峰基本一致。结论:仿制药A在4种溶出介质中的溶出行为与原研药一致;仿制药B在含0.6%十二烷基硫酸钠的水溶液中的溶出行为与原研药不同;原研药掰分前后批内均一性无明显变化,而仿制药A、B掰分后的批内均一性较整片有所降低;仿制药的脆碎度和分割质量损失均高于原研药;两者原料药晶型相同但晶体形貌存有差异。
关键词 奥卡西平;溶出行为;刻痕片;仿制药;原研药;一致性评价;高效液相色谱法
Consistency Evaluation on the Dissolution Behavior and Related Indicators between Generic and Original Preparations of Oxcarbazepine Scored Tablets
LYU Beibei1,2,YANG Haiyuan1,2,CHENG Hua1,2,WEI Wenzhi1,2,ZHANG Minjuan1,2(1. Chemistry Laboratory, Qinghai Institute for Drug Control, Xining 810016, China; 2. Qinghai Provincial Key Laboratory of Modernization of Traditional Chinese and Tibetan Medicine, Xining 810016, China)
ABSTRACT OBJECTIVE: To evaluate the dissolution behavior consistency between the generic drugs and original drugs of Oxcarbazepine scored tablets, and to compare the appearance, the friability of the split portions, loss of mass of the split portions as well as crystal form and morphology of raw material from different enterprises. METHODS: HPLC method was adopted. The paddle method (rotation speed of 60 r/min, the temperature of 37.0℃) was adopted to determine accumulative dissolution rate of generic and original drugs in 4 mediums [0.6% SDS hydrochloric acid solution (pH=1.2), 0.6% SDS acetate buffer solution (pH=4.5), 0.6% SDS phosphate buffer solution (pH=6.8) and 0.6% SDS water solution]. The similarity factor method was used to evaluate the similarity of dissolution curves as well as intra-batch uniformity of the split portions and whole tablets. The friability tester and electronic balance were used to determine the friability and the loss of mass of the split portions. X-ray diffractometer and scanning electron microscope were used to observe the crystal form and crystal morphology of the raw materials of different enterprises. RESULTS: The linear range of oxcarbazepine was 33.42-401.09 μg/mL(r=0.999 9);LOQ was 0.10 μg/mL, and LOD was 0.04 μg/mL; RSDs of precision,stability, reprodu- cibility and durability tests were lower than 2.0%; the reco- veries were 99.80%-101.63% (RSD=0.37%-0.91%,n=3). The average cumulative dissolution rate of generic drug A, generic drug B and original drug in 4 different dissolution media at 90 min were 92%, 87%, 90% [0.6% SDS hydrochloric acid solution (pH=1.2)];94%, 94%, 90% [0.6% SDS acetate buffer solution (pH=4.5)];95%, 95%, 91% [0.6% SDS phosphate buffer solution (pH 6.8)]; 97%, 98%, 95% (0.6% SDS water solution). The similarity factors of generic drug A, generic drug B and original drug in 4 kinds of different dissolution media were 66 and 81,71 and 69,71 and 61,59 and 39. In the first 15 min, the difference of dissolution rate of split portions and whole tablets were -3%-13%, -2%-24% and -3%-7% for generic drug A, generic drug B and original drug, respectively. RSDs of accumulative dissolution rate of split portions and whole tablets were 6%-14% and 2%-9% for generic drug A (n=12), 4%-10% and 1%-8% for generic drug B (n=12) and 2%-7% and 2%-8% for original drug. The appearance of the original drug was fusiform, and the notch was deep;the shape of the generic drug was different from each other, and the notch of the generic drug was significantly shallower than that of original drug. The friability, the loss of mass of the split portions for generic drug A and generic drug B, original drug were 0.62%and 0.67%,0.12% and 0.11%,0.08% and 0.05%. The domestic raw materials possessed irregular lumps and debris, while the raw materials produced by original drug enterprises possessed regular flat cuboids and regular strips with little debris; but X-ray diffraction peaks of them were basically the same. CONCLUSIONS: The dissolution behavior of generic drug A in 4 medium is consistent with that of the original drug; dissolution behavior of generic drug B in water containing 0.6%SDS is different from that of the original drug; there is no significant change in the homogeneity of the original drug before and after splitting, but the homogeneity of the generic drug A and B after splitting is lower than that of the whole tablet; the fragility of generic drugs and loss of mass of split portions are higher than those of the original drugs; two kinds of raw material have the same crystal form but different crystal morphology.
KEYWORDS Oxcarbazepine; Dissolution behavior; Scored tablets; Generic drug; Original drug; Consistency evaluation; HPLC
功能性刻痕片剂(可分割的刻痕片)是一种带有一道或多道刻痕,以便于进行剂量分割的片剂[1]。其具有灵活调整药物剂量、弥补儿童或老年用药规格不足、降低患者用药成本的特点,但存在分剂量不均匀[2-3]、分割时出现粉末或碎片导致药量损失的现象[4]。分剂量不均匀会影响药物的疗效或引发不良反应,特别是对于治疗窗窄的药物[5-6]。9.0版《欧洲药典》[7]、41版《美国药典》[8]均有相应的关于刻痕片的技术要求和规范,而《中国药典》暂未收录相关内容。美国FDA于2013年发布了相关技术指南[9],进一步对刻痕片剂进行了管理和规范。我国国家药品监督管理局药品审评中心(CDE)网站于2019年发布了《仿制口服片剂功能性刻痕设计和研究的一般要求》[1],要求仿制药的功能性刻痕应该与原研药保持一致,且须对分割后的制剂单元进行质量差异或含量均匀度、分割质量损失、脆碎度、溶出度等进行考察。功能性刻痕片作为口服固体制剂,溶出度是其关键质控属性之一[10]。采用溶出曲线测定原研药与仿制药的溶出度,既可为仿制药研发过程中处方的筛选提供参考,又可作为仿制药质量与疗效一致性评价的重要手段[11]。
奥卡西平(Oxcarbazepine)是一种新型抗癫痫药物,为卡马西平的10-酮基衍生物,主要通过其活性代谢产物10-单羟基代谢物发挥作用,具有耐受性好、毒副作用小、药物间相互作用少等特点,已成为治疗成人及儿童癫痫局部发作或全身强直性阵挛发作的一线药物[12]。奥卡西平刻痕片原研药可掰分为两个半片,极大地弥补了儿童用药规格不足的情况[13]。
本研究通过高效液相色谱法(HPLC)结合相似因子法比较了不同溶出介质中奥卡西平仿制药与原研药的溶出曲线,并对掰分后的半片制剂进行了溶出曲线考察,以剖析奥卡西平刻痕片整片制剂与半片制剂的溶出行为;同时,本研究还比较了全片外观,半片制剂的分割质量损失、脆碎度及原料药的晶型、晶体形貌,旨在评价仿制药与原研藥的一致性,并为仿制药的生产工艺和内在质量提升提供参考,亦为刻痕片的质量控制提供依据。
1 材料
1.1 仪器
1260型HPLC仪,配备G7111A型四元泵、G7129A型自动进样器、G7116A型柱温箱、G71157型二极管阵列检测器(美国Agilent公司);AT型全自动溶出仪(瑞士Sotax公司);FAVD-25型真空脱气仪(上海富斯科分析仪器有限公司);PB-21型酸度计、CP225D型十万分之一电子天平(德国Sartorious公司);H1850R型高速冷冻离心机(湖南长沙湘仪离心机仪器公司);D8 Advance型X射线衍射仪(德国Bruker公司);JSM-6390型扫描电子显微镜(日本JEOL公司);FT-2000型脆碎度检测仪(天津市矽新科技有限公司);KQ-500DE型数控超声波清洗器(昆山市超声仪器有限公司)。
1.2 药品与试剂
奥卡西平对照品(中国食品药品检定研究院,批号:100657-201102,纯度:99.8%);奥卡西平刻痕片原研药(瑞士Novartis公司,以下简称“企业R”,规格:300 mg,批号:TR228、T2506、TU899);仿制药A(国内A企业,规格:300 mg,批号:180411、180109、190103);仿制药B(国内B企业,规格:300 mg,批号:181109、190101、171003);奥卡西平原料药1(国内企业1,批号:180602,纯度:99.4%);奥卡西平原料药2(国内企业2,批号:170620,纯度:99.2%);奥卡西平原料药3[北京诺华制药有限公司(即原研药企业中国分公司)赠送,批号:181105,纯度:99.6%];十二烷基硫酸钠(SDS)、乙腈均为色谱纯,其余试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:Thermo BDS Hypersil C18(150 mm×4.6 mm,5 μm);流动相:乙腈-0.05 mol/L磷酸二氢钾水溶液(含0.2%三乙胺,用磷酸调节pH至6.0)(40 ∶ 60,V/V);检测波长:256 nm;流速:1.0 mL/min;柱温:30 ℃;进样量:10 μL。
2.2 溶出介质的选择与配制
在2015年版《中国药典》(二部)[14]、美国FDA橙皮书数据库[15]、41版《美国药典》[8]收录的奥卡西平片的质量标准中,均在溶出介质中加入了SDS(每300 mg的质量百分比均为0.6%)以提高药物的溶解度。为考察溶液pH对奥卡西平溶解性能的影响,本研究分别测定奥卡西平原料药(批号:180602)在含0.6%SDS的盐酸溶液(pH=1.0、pH=2.0)、含0.6%SDS的醋酸盐缓冲溶液(pH=4.5)、含0.6%SDS的磷酸盐缓冲溶液(pH=6.0、pH=6.8)、含0.6%SDS的水溶液中的溶解度。结果发现,奥卡西平在不同pH条件下的溶解度均在0.3 mg/mL左右,说明pH对奥卡西平溶解度无明显影响。根据《普通口服固体制剂溶出度试验技术指导原则》[16],本研究最终选择了含0.6%SDS的盐酸溶液(pH=1.2)、含0.6%SDS的醋酸盐缓冲溶液(pH=4.5)、含0.6%SDS的磷酸盐缓冲溶液(pH=6.8)、含0.6%SDS的水溶液为溶出介质,并参照《普通口服固体制剂溶出曲线测定与比较指导原则》[17]中溶出介质配制方法进行配制。
2.3 溶液的制备
2.3.1 对照品溶液 精密称取奥卡西平对照品33 mg,置于100 mL量瓶中,加乙腈10 mL,超声(功率:500 W,频率:40 kHz,下同)使溶解,分别用4种溶出介质稀释至刻度,即得4种质量浓度均为330 μg/mL的对照品溶液。
2.3.2 供试品溶液 ①按2015年版《中国药典》(四部)通则“0931第二法(桨法)”[18]分别取“2.2”项下4种溶出介质900 mL,置于溶出杯中。取9批样品,每批12片,分别置于上述溶出杯中,设置温度37.0 ℃、转速60 r/min,分别于5、10、15、20、30、45、60、90 min时取溶液5 mL,再经自动溶出仪自动补加相应溶出介质5 mL;取样溶液经0.45 μm滤膜滤过,取续滤液,即得。②取样品适量,精密称定,置于500 mL量瓶中,加入含0.6%SDS的水溶液适量,振摇45 min,用含0.6%SDS的水溶液稀释至刻度,摇匀,滤过,取续滤液,即得。③取样品适量,精密称定,分别置于100 mL量瓶中,加乙腈10 mL,超声使溶解,用含0.6%SDS的水溶液稀释至刻度,摇匀,滤过,取续滤液,即得。
2.3.3 空白对照溶液 参照辅料成分较多的仿制药A的处方工艺,按比例称取各辅料适量,混匀,加入含0.6%SDS的水溶液900 mL,超声10 min,经0.45 μm滤膜滤过,取续滤液,作为空白对照溶液。
2.4 含量测定
2.4.1 专属性试验 取“2.2”项下4种溶出介质、“2.3.3”项下空白对照溶液、“2.3.1”项下4种对照品溶液及“2.3.2①”项下4种供试品溶液各适量,按“2.1”项下色谱条件进样测定,记录色谱图。结果显示,4种溶出介质和空白对照对测定均无干扰,奥卡西平和相邻杂质的分离度均大于1.5,理论板数按奥卡西平计均不低于3 000,详见图1。
2.4.2 线性关系考察 精密称取奥卡西平对照品83.56 mg,置于50 mL量瓶中,加乙腈30 mL,超声使溶解,加水稀释至刻度,得质量浓度为1 671.20 μg/mL的对照品贮备液。分别精密量取上述对照品贮备液0.5、1.0、2.0、3.0、4.0、5.0、6.0 mL,置于25 mL量瓶中,用含0.6%SDS的水溶液稀释至刻度,摇匀,制得系列线性对照品溶液,按“2.1”项下色谱条件进样测定,记录峰面积。以待测成分质量浓度(x,μg/mL)为横坐标、峰面积(y)为纵坐标进行线性回归,得奥卡西平的回归方程为y=16.182x+30.237(r=0.999 9)。这表明,奥卡西平检测质量浓度的线性范围为33.42~401.09 μg/mL。
2.4.3 定量限与检测限考察 参考预试验结果取“2.4.2”项下质量浓度为33.42 μg/mL的线性对照品溶液,精密量取1.0 mL,置于100 mL量瓶中,用含0.6%SDS的水溶液稀释至刻度,摇匀,精密量取3.0 mL,置于10 mL量瓶中,用含0.6%SDS的水溶液稀释至刻度,摇匀,按“2.1”项下色谱条件进样测定,以信噪比为10 ∶ 1计算得奥卡西平的定量限为0.10 μg/mL;取上述信噪比为10 ∶ 1的对照品溶液,精密量取4.0 mL,置于10 mL量瓶中,用含0.6%SDS的水溶液稀释至刻度,摇匀,按“2.1”项下色谱条件进样测定,以信噪比为3 ∶ 1计算得奥卡西平的检测限为0.04 μg/mL。
2.4.4 精密度试验 取“2.3.1”项下对照品溶液(以0.6%SDS的水溶液为溶剂),按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,奥卡西平峰面积的RSD为0.2%(n=6),表明仪器精密度良好。
2.4.5 稳定性试验 取“2.3.2①”项下溶出90 min时的4种供试品溶液各适量(仿制药A,批号:180109),分别于室温下放置0、2、4、6、8、10、12、14、16、18、20、24 h时,按“2.1”项下色谱条件进样测定,记录峰面积。结果,当以含0.6%SDS的盐酸溶液(pH=1.2)、含0.6%SDS的醋酸盐缓冲溶液(pH=4.5)为溶出介质时,供试品溶液放置10 h内峰面积的RSD分别为1.9%、1.7%(n=6),12~24 h内峰面积的RSD分别为2.2%、2.4%(n=7);当以含0.6%SDS的磷酸盐缓冲溶液(pH=6.8)、含0.6%SDS的水溶液为溶出介质时,供试品溶液放置24 h内峰面积的RSD分别为0.5%、0.4%(n=12)。这表明,当以含0.6%SDS的盐酸溶液(pH=1.2)、含0.6%SDS的醋酸盐缓冲溶液(pH=4.5)为溶出介质时,供试品溶液放置10 h内稳定性良好,当以含0.6%SDS的磷酸盐缓冲溶液(pH=6.8)、含0.6%SDS的水溶液為溶出介质时,供试品溶液放置24 h内稳定性良好。
2.4.6 重复性试验 取仿制药A(批号:180109)粉碎,取细粉约200 mg(约相当于奥卡西平150 mg),共6份,按“2.3.2②”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并按外标法计算样品中奥卡西平的含量。结果,奥卡西平平均含量的RSD为1.2%(n=6),表明方法重复性良好。
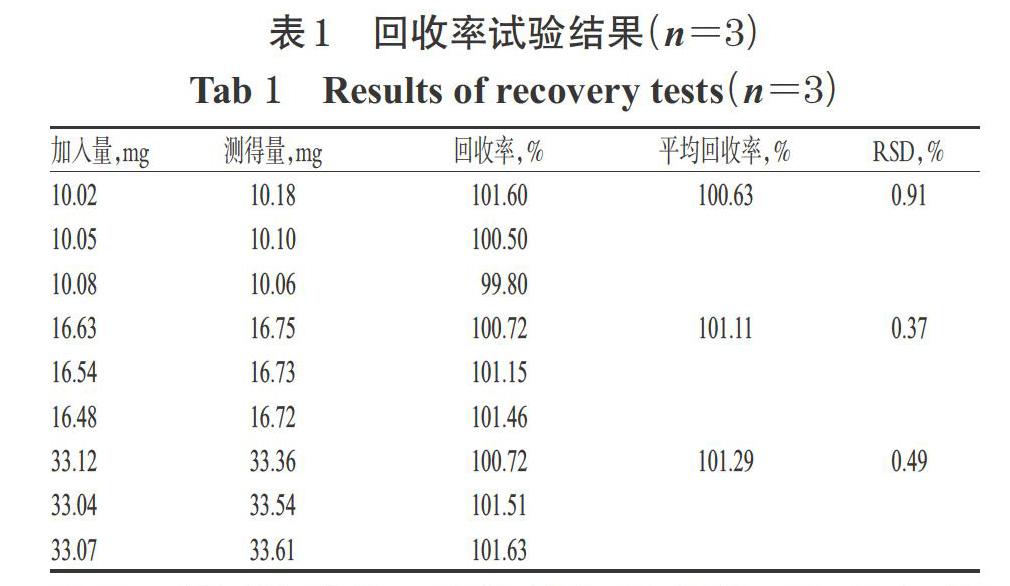
2.4.7 回收率试验 按仿制药A处方量的30%、50%、100%分别精密称取奥卡西平对照品适量,并按其处方工艺称取辅料,共9份,分别置于100 mL量瓶中,加乙腈10 mL,超声使溶解,用含0.6%SDS的水溶液稀释至刻度,摇匀,按“2.1”项下色谱条件进样测定,记录峰面积并计算回收率,结果见表1。
2.4.8 耐用性试验 取仿制药A(批号:180109),按“2.3.2①”项下方法制备供试品溶液(90 min),再按“2.1”项下色谱条件,分别以不同色谱住[Agilent Zorbax SB C18(150 mm×4.6 mm,5 μm)、Waters SunFire C18(150 mm×4.6 mm,5 μm)、Thermo BDS Hypersil C18(150 mm×4.6 mm,5 μm)]、HPLC仪(e2695型、1260型、LC-20AT型)、柱温(25、30、35 ℃)]进样测定,记录峰面积并按外标法计算样品含量。结果,奥卡西平含量的RSD均小于2.0%(n=3),表明该方法能够满足试验要求,耐用性良好。
2.4.9 样品含量测定 取9批样品细粉,每份约40 mg(约相当于奥卡西平30 mg),按“2.3.2③”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并按外标法计算样品含量,平行操作2次。结果,仿制药A、B及原研药的平均含量分别为100.2%、99.9%、99.6%。
2.5 滤膜吸附性考察
以0.6%SDS的水溶液为溶出介质,取仿制药A(批号:180109)适量,按“2.3.2①”项下方法制备供试品溶液,取90 min时的供试品溶液150 mL,置于500 mL量瓶中,用含0.6%SDS的水溶液稀释至刻度(约相当于第1个取样时间点奥卡西平的质量浓度),摇匀,作为滤膜吸附性考察溶液。取上述滤膜吸附性考察溶液适量,以 4 000 r/min离心10 min,取上清液,作为离心样品;另取滤膜吸附性考察溶液,分别用滤膜(0.45 μm)依次滤过3、5、7、10 mL,作为滤膜滤过样品。同时为考察自动溶出仪取样管路是否存在吸附性,分别采用手动取样和仪器自动取样的方式取样,按上述方法分别滤过5、10 mL滤膜吸附性考察溶液,作为手动取样滤过样品和自动取样管路滤过样品。取上述各样品适量,分别按“2.1”项下色谱条件进样测定,记录峰面积并计算吸附率:吸附率(%)=(1-滤过样品峰面积/离心样品峰面积)×100%。结果,3、5、7、10 mL滤膜滤过样品的吸附率分别为0.7%、0.3%、0.5%、0.2%,5、10 mL手动滤过样品的吸附率分别为0.2%、0.3%,5、10 mL自动取样吸附率分别为0.4%、0.1%(均小于2.0%),表明滤膜、自动取样管路对样品均无吸附性。
2.6 溶出行为考察
2.6.1 仿制药与原研药(整片)的溶出度 取9批样品,每批12片,按“2.3.2①”项下方法考察其在4种溶出介质中的溶出行为,并以溶出时间为横坐标(x)、各时间点样品累积溶出度的平均值为纵坐标(y),绘制溶出曲线[累积溶出度(%)=An+[(An-1+…A2+A1)×V1
V2] ,式中V1为各时间点固定取样体积,V2为溶出介质体积,其中An为各时间点测得溶出度[19]]。结果,仿制药与原研药整片制剂的溶出曲线在以含0.6%SDS的水溶液中存在较大差异,其中原研药和仿制药A、B在10 min时的累积溶出度分别为77%、66%、48%,说明原研药在该时间点的溶出速度明显快于仿制药。原研药和仿制药A、B在其余3种溶出介质中10 min时的累积溶出度分别为68%、65%、64%[含0.6%SDS的盐酸溶液(pH=1.2)];71%、64%、69%[含0.6%SDS的醋酸盐缓冲溶液(pH=4.5)];72%、65%、62%[含0.6%SDS的磷酸盐缓冲溶液(pH=6.8)],提示3种样品在其余3种溶出介质中的溶出行为无明显差异。原研药和仿制药A、B在含0.6%SDS的盐酸溶液(pH=1.2)中累积溶出度超过85%所需的时间分别为45、30、45 min;在其余3种溶出介质中,不同企业样品的累积溶出度在30 min时均达到85%以上。仿制药A、B和原研药在4种不同溶出介质中90 min时的平均累积溶出度分别为92%、87%、90%[含0.6%SDS的盐酸溶液(pH=1.2)],94%、94%、90%[含0.6%SDS的醋酸盐缓冲溶液(pH=4.5)],95%、95%、91%[含0.6%SDS的磷酸盐缓冲溶液(pH=6.8)],97%、98%、95%(含0.6%SDS的水溶液),详见图2。
式中Rt为t时点原研药的平均累积溶出度,Tt为t时点仿制药的平均累积溶出度,n为取样时点的个数[17]。结果显示,仿制药A、B与原研药在含0.6%SDS的盐酸溶液(pH=1.2)、含0.6%SDS的醋酸盐缓冲溶液(pH=4.5)、含0.6%SDS的磷酸盐缓冲溶液(pH=6.8)、含0.6%SDS的水溶液中的相似因子分别为66、81,71、69,71、61,59、39。表明仿制药A在4种溶出介质中的溶出曲线与原研药相似,而仿制药B在含0.6%SDS的水溶液中的溶出曲线与原研药存在差异,提示含0.6%SDS的水溶液具有区分力。
2.6.2 仿制药与原研药(半片)的溶出度及其差异 根据国家CDE网站发布的《仿制口服片功能性刻痕设计和研究的一般要求》,刻痕片分割后的部分的溶出度应符合成品质量标准[1]。2015年版《中国药典》(二部)中记载奥卡西平片在含0.6%SDS的水溶液中30 min时的溶出度应不低于75%,因此掰分后的半片制剂在含0.3%SDS的水溶液中30 min時的溶出度也应不低于75%[14]。本研究按“2.6.1”项下方法考察了半片制剂的溶出行为。结果,仿制药A、B及原研药掰分后的半片制剂在上述条件下的溶出度分别为95%、93%、95%,均符合要求。为进一步考察奥卡西平刻痕片掰分后的溶出行为变化,取9批样品,每批12片,掰成半片,考察其在4种溶出介质中与整片制剂的溶出行为差异。结果,前15 min内,仿制药A、B的半片制剂在4种溶出介质中的溶出度均显著升高,较原研药差异明显,其中仿制药B半片与整片制剂的溶出度差值范围最大,为-2%~24%,仿制药A为-3%~13%,原研药为-3%~7%,详见图3(因180401、190101、TR228批样品的半片制剂在5 min时溶出度的RSD超过20%,故剔除5 min时间点的数据,下同)。
2.6.3 半片与整片制剂的批内均一性 为考察不同企业样品掰分后的溶出均一性变化,本研究按2015年版《中国药典》(二部)中奥卡西平片的溶出介质(整片制剂为含0.6%SDS的水溶液,半片制剂为含0.3%SDS的水溶液),随机选取不同企业的1批样品(仿制药A批号:180109、仿制药B批号:190101、原研药批号:TU899),每批12片,按“2.3.2①”项下方法制备供试品溶液,以累积溶出度的RSD为指标,考察同一批样品半片与整片的批内均一性(剔除5 min时间点)。结果,仿制药A半片与整片各时间点累积溶出度的RSD分别为6%~14%、2%~9%(n=12),仿制药B为4%~10%、1%~8%(n=12),原研药为2%~7%、2%~8%(n=12)。这表明,原研药掰分前后批内均一性无明显变化,而仿制药A、B掰分后批内均一性较整片有所降低。
2.7 外观
奥卡西平原研药外观为梭形,刻痕较深;仿制药片形各异,呈圆形或胶囊型,刻痕明显浅于原研药。采用手掰的方式进行分割后发现,仿制药掰分时比较费力且掰分后不均匀;原研药易于操作,掰分后均匀整齐。
2.8 脆碎度及分割质量损失考察
取9批样品,每批12片,采用脆碎度检测仪及十万分之一电子天平测定掰分后半片制剂的脆碎度和分割质量损失。结果,原研药的平均脆碎度、分割质量损失分别为0.08%、0.05%,仿制藥A分别为0.62%、0.67%,仿制药B分别为0.12%、0.11%,均符合相关要求[1],但仿制药的脆碎度和分割质量损失均高于原研药。
2.9 原料药的晶型与晶体形貌比较
有文献报道,奥卡西平粉末X射线衍射的特征峰位于7.236°、10.220°、11.977°、14.399°、17.700°、19.098°、19.457°、20.060°、21.120°、21.978°、23.040°、23.658°、25.200°、25.637°、26.141°处[20]。本研究参照上述文献对不同企业奥卡西平原料药(原料药1~3)进行X射线衍射测定。结果,3种原料药X射线衍射图谱几乎完全相同,特征峰与文献报道[20]基本一致,表明3种原料药晶型一致(图4)。将3种原料药进行电镜扫描发现,原料药1、2的晶体形貌呈不规则的块状和碎屑,原料药3呈规则的扁平长方体和规则的条状且碎屑少(图5)。这表明,国产原料药与原研药企业产原料药的晶体形貌有所不同。
3 讨论
2015年版《中国药典》(二部)中以乙腈-0.05 mol/L磷酸二氢钾水溶液(用磷酸调节pH至3.0)(40 ∶ 60,V/V)为流动相[14]。但笔者在试验过程中发现,当水相pH为3.0时,对照品溶液在紧邻主峰前出现一小峰,无法基线分离;且在调整流动相比例以及更换不同色谱柱后均无明显改善。但调整水相的pH(pH=4.0、5.0、6.0)且当其pH为6.0时,该小峰消失,更换不同色谱柱以及不同品牌HPLC仪后也未再出现。有文献报道,有些样品在特定的色谱条件下可能存在结构的动态平衡而出现双峰,这种双峰是无法完全分离的,当改变色谱条件尤其是pH值时会使峰形正常[21]。因此,有必要充分考虑待分离样品的酸度系数(pKa),以保证样品在流动相pH条件下能以分子状态存在[22]。由于奥卡西平的pKa为13.73[23],存在酰胺键,当流动相酸性较强时有可能出现上述文献所述的动态结构平衡,故笔者调整水相pH后双峰现象消失。
笔者在前期预试验中发现,按“2.3.2①”项下方法制备供试品溶液进行重复性试验时,奥卡西平含量的RSD值较大。进行含量测定时,样品不能完全溶解,测得的含量较低,故本研究采用了不同的方法制备供试品溶液。
刻痕线的形状、深度,刻痕片的形状、大小、厚薄及曲率均可能影响片剂分剂量的准确性[1]。本研究结果显示,仿制药掰分时较费力且存在分剂量不均匀的现象,该结果与仿制药半片制剂溶出均一性降低结果一致;仿制药的脆碎度和分割质量损失均高于原研药,表明仿制药与原研药的工艺参数可能存在差异[24],提示生产工艺可影响掰分后分剂量的准确性及溶出行为。
通过对不同企业奥卡西平原料药进行比较后发现,国产原料药与原研药企业产原料药虽然晶型相同,但晶体形貌有所不同。这不仅可能对粒子的表面积及粒子间的相互作用产生影响,还可能影响辅料混合以及制粒压片,从而影响药物的溶出度[25-26]。因此,对于难溶性药物除需关注晶型[27]对药物溶出度的影响外,还应当重视晶体形貌对溶出行为的影响。
综上所述,奥卡西平刻痕片原研药和仿制药在溶出行为及均一性等指标上均存有一定差异,仿制药的生产工艺和内在质量有待进一步提升。
参考文献
[ 1 ] 国家药品监督管理局药品审评中心.仿制口服片剂功能性刻痕设计和研究的一般要求[EB/OL].(2019-07-04)
[2019-09-24]. http://www.cde.org.cn/news.do?method=
viewInfoCommon&id=314888.
[ 2 ] SHAH RB,COLLIER JS,SAYEED VA,et al. Tablet splitting of a narrow therapeutic index drug:a case with levothyroxine sodium[J]. AAPS PharmSciTech,2010,11(3):1359-1367.
[ 3 ] ROSENBERG JM,NATHAN JP,PLAKOGIANNIS F. Weight variability of pharmacist-dispensed split tablets[J].J Am Pharm Assoc:Wash,2002,42(2):200-205.
[ 4 ] FREEMAN MK,WHITE W,IRANIKHAH M. Tablet splitting:a review of weight and content uniformity part 1 of a 2-part series:next month:table splitting:a review of the clinical and economic outcomes and patient acceptance[J]. Consult Pharm,2012,27(5):341-352.
[ 5 ] 商国美,赵荣伟.我院住院处方最小规格剂量单位再分零使用情况分析[J].西北药学杂志,2005,20(4):175-176.
[ 6 ] 吴婧倩,杨颖昕,吕志杰.我院住院药房分劈药片使用状况调查及持续质量改进[J].实用药物与临床,2018,21(2):235-237.
[ 7 ] European Pharmacopoeia Commission. European pharmacopeia 9.0[S]. Strasbourg:European Directorate for the Quality of Medicine,2017:885.
[ 8 ] The United States Pharmacopeial Convention. The United States pharmacopeia 41:volume 2[S]. Baltimore:United Book Press,2018:3080、6457.
[ 9 ] FDA. Tablet scoring:nomenclature,labeling,and data for evaluation[EB/OL].(2013-03-13)[2019-06-04]. https://www.fda.gov/drugs/guidances-drugs/all-guidances-drugs.
[10] 金方方,尹婕,南楠.化学口服固体制剂仿制药质量和疗效一致性评价研究思考[J].药物分析杂志,2018,38(4):575-581.
[11] 谢沐风.改善溶出度评价方法,提高固体药物制剂水平[J].中国医药工业杂志,2005,36(7):447-451.
[12] 丁晶,汪昕.癫痫诊疗指南解读[J].临床内科杂志,2016,33(2):142-144.
[13] 胡利华,王晓玲.我院片剂分剂量使用现状调查分析[J].儿科药学杂志,2013,19(3):32-35.
[14] 国家药典委员会.中华人民共和国药典:二部[S]. 2015年版.北京:中国医药科技出版社,2015:1406.
[15] FDA. Drug database:dissolution methods[EB/OL]. (2004-
02-12)[2019-06-04]. https://www.accessdata.fda.gov/scripts/cder/dissolution/index.cfm.
[16] 国家食品药品监督管理总局.普通口服固体制剂溶出度试验技术指导原则[EB/OL].(2015-02-05)[2019-06-04].
http://www.sfda.gov.cn/WS01/CL0087/114286.html.
[17] 国家食品药品监督管理总局.普通口服固体制剂溶出曲线测定与比较指导原则[EB/OL].(2016-03-18)[2019-06-
05].http://www.sfda.gov.cn/ws0l/cl0087/147583.html.
[18] 国家药典委员会.中华人民共和国药典:四部[S]. 2015年版.北京:中国医药科技出版社,2015:121.
[19] 张启明,谢沐风,宁保明,等.采用多条溶出曲线评价口服固体制剂的内在质量[J].中国医药工业杂志,2009,40(12):946-955.
[20] 梁忠瑞.奥卡西平共晶的制备、表征与性质研究[D].北京:北京化工大学,2018.
[21] 龚时琼.高效液相色谱分析中异常峰的分析与处理[J].实验技术与管理,2010,27(6):37-42.
[22] 全红娜,金松子,雷勇生,等.反相高效液相色谱中流动相选择与优化的研究进展[J].现代药物与临床,2014,29(10):1190-1194.
[23] FDA. Approved drugs:oxcarbazepine[EB/OL].(2018-10-
13)[2019-06-04]. https://pdf.hres.ca/dpd_pm/00048247.
[24] 王芳,金曼,朱传祥.影响片剂脆碎度的因素分析[J].齐鲁药事,2012,31(10):613-614.
[25] 申宝德,沈成英,徐玲霞,等.难溶性中药纳米混悬剂的体内外行为研究进展[J].中国中药杂志,2018,43(19):3828-3833.
[26] 金瑞,程开生,李志云,等.缬沙坦原料颗粒形貌对缬沙坦氨氯地平片溶出的影响[J].中国粉体技術,2019,25(5):45-50.
[27] 谭菊英,黄丽丽,孙煜,等.吡罗昔康原料晶型对片剂溶出度的影响研究[J].药物分析杂志,2017,37(3):550-557.
(收稿日期:2020-01-09 修回日期:2020-05-08)
(编辑:陈 宏)
