腰痛康胶囊的质量标准研究
2017-01-03徐倩邱慧
徐倩+邱慧
[摘要]目的 建立腰痛康胶囊的质量标准。方法 采用薄层色谱法(TLC)对处方中续断、独活、狗脊等药材进行薄层鉴别;反相高效液相色谱法(HPLC)测定腰痛康胶囊中淫羊藿苷的含量。结果 TLC中斑点清晰、分离良好;HPLC测定淫羊藿苷进样量在0.21~2.1 μg范围内线性关系良好(r=0.9984),平均加样回收率(n=5)为99.2%,RSD(n=5)为0.25%。结论 本方法操作简便、专属性强、准确度高、精密度和重复性好,可作为该制剂的质量控制标准。
[关键词]质量标准;腰痛康胶囊;薄层色谱法;高效液相色谱法
[中图分类号] R927.2 [文献标识码] A [文章编号] 1674-4721(2016)10(b)-0007-05
[Abstract]Objective To establish the quality standards of Yaotongkang Capsules.Methods Radix dipsaci,angelica wolfberry,rhizoma etc. medicinal materials in the prescription were identified by using thin layer chromatography (TLC);content of epimedium glycoside in Yaotongkang Capsules was detected by reverse phase high performance liquid chromatography (HPLC) method.Results TLC spots were clear and well separated;HPLC for sample size of epimedium glycoside in the range of 0.21-2.1 μg was good (r=0.998 4),average recovering rate was 99.2% (n=5),RSD was 0.25% (n=5).Conclusion The method has the advantages of simple operation,strong specificity,high accuracy,good precision and repeatability,and can be used as the quality control standard of the preparation.
[Key words]Quality standards;Yaotongkang Capsules;Thin layer chromatography;High performance liquid chromatography
腰痛康胶囊为南昌市洪都中医院自制制剂,处方来源于该院骨伤科名老中医根据祖国传统中医理论,结合多年临床经验总结而成的,处方由淫羊藿、桑寄生、续断、狗脊、鹿角霜、牛膝、独活、生姜等15味中药组成,具有补肝益肾、理气通络、活血止痛的功效,用于肝肾亏虚、寒湿遇阻所致的急慢性腰痛、腰腿疼痛等症。为全面地控制药品质量,本文首次对腰痛康胶囊的质量标准进行了研究。淫羊藿为处方中君药,味辛甘性温,走肝肾二经,为补命门、强筋骨、益精气、温肾壮阳之要药[1]。淫羊藿苷作为淫羊藿的重要活性成分[2],其含量测定多采用高效液相色谱法(high performance liquid chromatography,HPLC)[3-5]。本研究选择淫羊藿苷为研究目标进行腰痛康胶囊的含量测定试验。
1仪器与试剂
Shimadzu LC-20AT系列高效液相色谱仪(日本岛津公司);Sartorius BS224S电子天平(赛多利斯科学仪器有限公司);Mettler-Toledo XS105 DualRange电子天平(梅特勒-托伦多称重设备系统有限公司);UV-2550紫外可见光分光光度计(日本岛津公司);KQ-300DB型超声波清洗器(昆山超声仪器有限公司);DKS-12电热恒温水浴锅(沈荡中新电器厂);202-2-S电热恒温干燥箱(上海跃进医疗器械厂)。
腰痛康胶囊7批,均由南昌市洪都中医院制剂中心提供,阴性样品按处方自制。淫羊藿苷对照品(批号:110737-200415)、川续断皂苷Ⅵ对照品(批号:111685-200401)、蛇床子素对照品(110822-200406)、原儿茶酸对照品(110809-201205),均购自中国食品药品检验检定研究院。D101型大孔树脂(上海蓝季,中国)、聚酰胺粉(上海谱振生物科技有限公司)、硅胶G(青岛海洋化工有限公司)、硅胶H(青岛海洋化工有限公司)。乙腈(色谱纯,Merck公司),水为超纯水(Purifier DZG-303A超纯水系统,博科公司),其余试剂均为分析纯。
2方法与结果
2.1淫羊藿的薄层鉴别
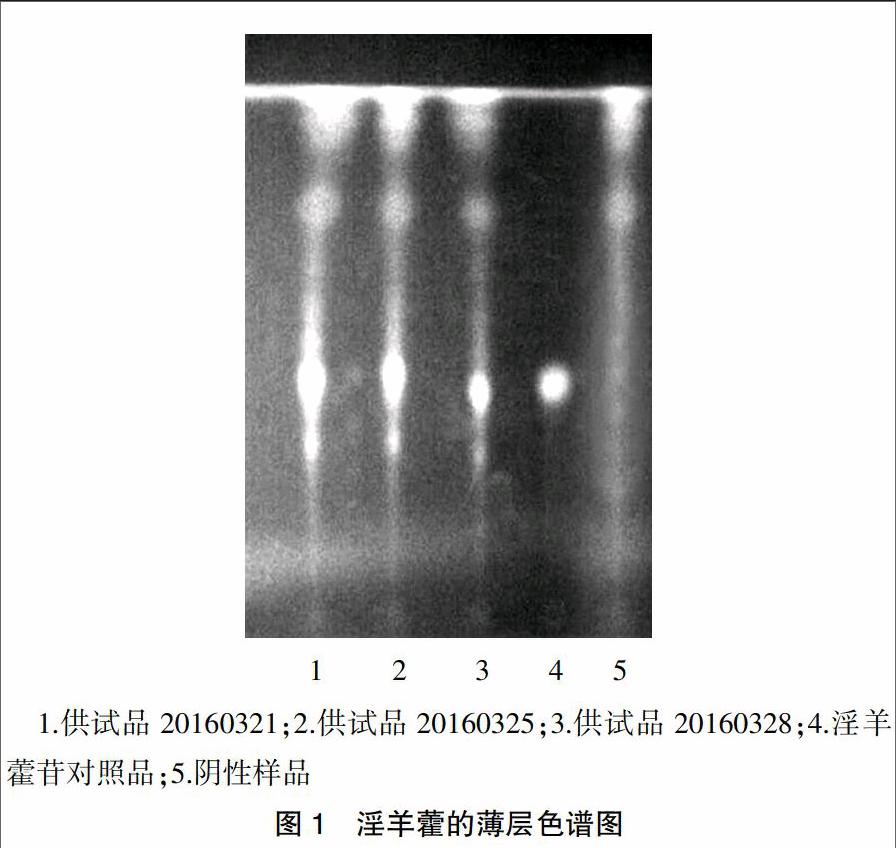
取本品内容物5 g,研细,加水30 ml使溶解,用乙醚提取3次,每次20 ml,水层挥尽乙醚,用乙酸乙酯提取2次,每次20 ml,合并乙酸乙酯液,蒸干,残渣加水20 ml使溶解,加至已处理好的聚酰胺柱(内径约1 cm,2 g,干法上柱)上,用乙酸乙酯50 ml洗脱,收集洗脱液并蒸干,残渣用1 ml甲醇溶解,作为供试品溶液。再取淫羊藿苷对照品,用甲醇溶解制成每1毫升含1 mg的溶液作为对照品溶液。为增加鉴别的专属性,取去除淫羊藿的模拟处方制成阴性样品,再按上述供试品溶液的制备方法依法制成缺淫羊藿的阴性样品溶液。按照薄层色谱法(thin layer chromatography,TLC)《中国药典》2015年版一部附录Ⅵ B试验,分别吸取供试品溶液、阴性样品溶液各15~20 μl以及对照品溶液5 μl,点于同一硅胶H薄层板上,展开剂为乙酸乙酯-丁酮-甲酸-水(10∶1∶1∶1),展开后取出,晾干并喷三氯化铝试液,于105℃烘干3~5 min,置于紫外光灯(365 nm)下检视。结果显示,供试品色谱与对照品色谱在相应的位置上显相同颜色的斑点(图1)。
2.2续断的薄层鉴别
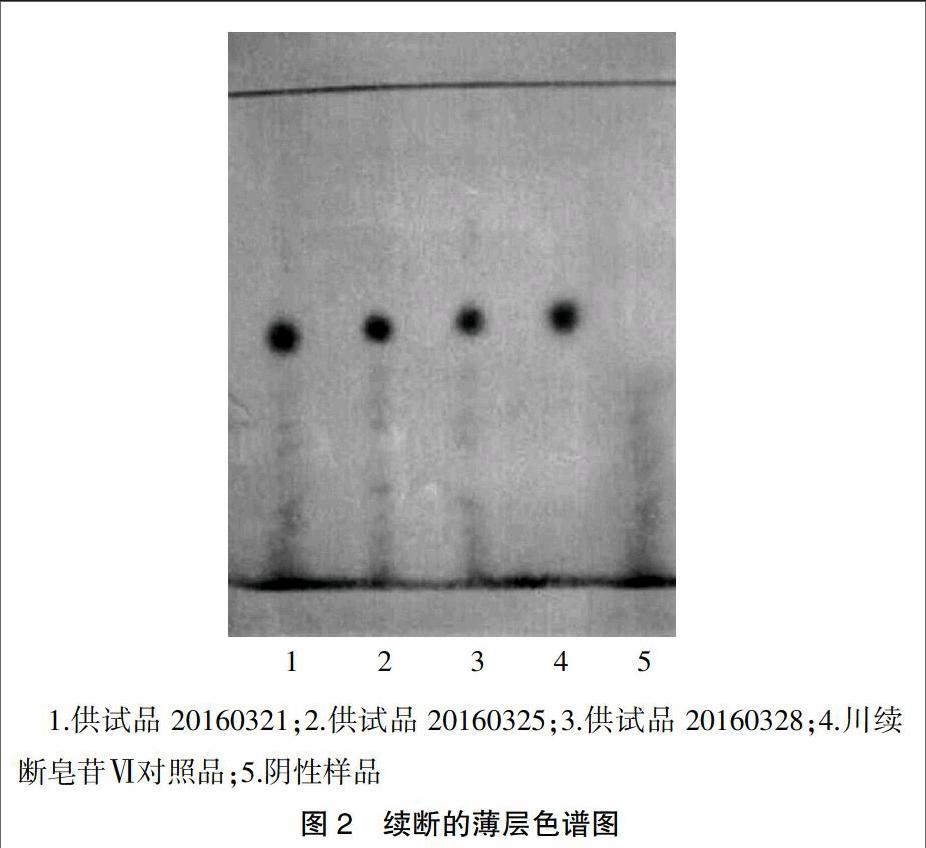
取本品内容物4.5 g,研细,置具塞锥形瓶中,移入甲醇25 ml,超声处理(300 W,25 kHz)30 min,滤过,滤液蒸干,残渣加水20 ml使溶解,用水饱和的正丁醇振摇提取3次,每次25 ml,合并正丁醇提取液,正丁醇提取液继续用氨试液洗涤2次,每次15 ml,弃去氨试液,再用水洗涤3次,每次30 ml,弃去水洗液,正丁醇液蒸干,残渣加水5 ml使溶解,上样于D101型大孔树脂柱(内径1.5 cm,长12 cm),依次用水、30%乙醇各50 ml进行洗脱,弃去洗脱液,再用70%乙醇50 ml洗脱,收集洗脱液后蒸干,用甲醇1 ml溶解残渣作为供试品溶液。再取川续断皂苷Ⅵ对照品,加甲醇溶解制成1 mg/ml的溶液,作为对照品溶液。为增加鉴别的专属性,取去除续断的模拟处方制成阴性样品,再按上述供试品溶液的制备方法依法制成缺续断的阴性样品溶液。照薄层色谱法《中国药典》2015年版一部附录Ⅵ B试验,分别吸取供试品溶液、阴性样品溶液各15~20 μl以及对照品溶液5 μl,点于同一硅胶G薄层板上,展开剂为三氯甲烷-甲醇-水(13∶7∶2)在10℃以下放置过夜的下层溶液,展开后取出晾干,并喷以10%硫酸乙醇溶液,于105℃加热至斑点清晰显色。结果显示,供试品色谱与对照品色谱在相应的位置上显相同颜色的斑点(图2)。
2.3独活的薄层鉴别
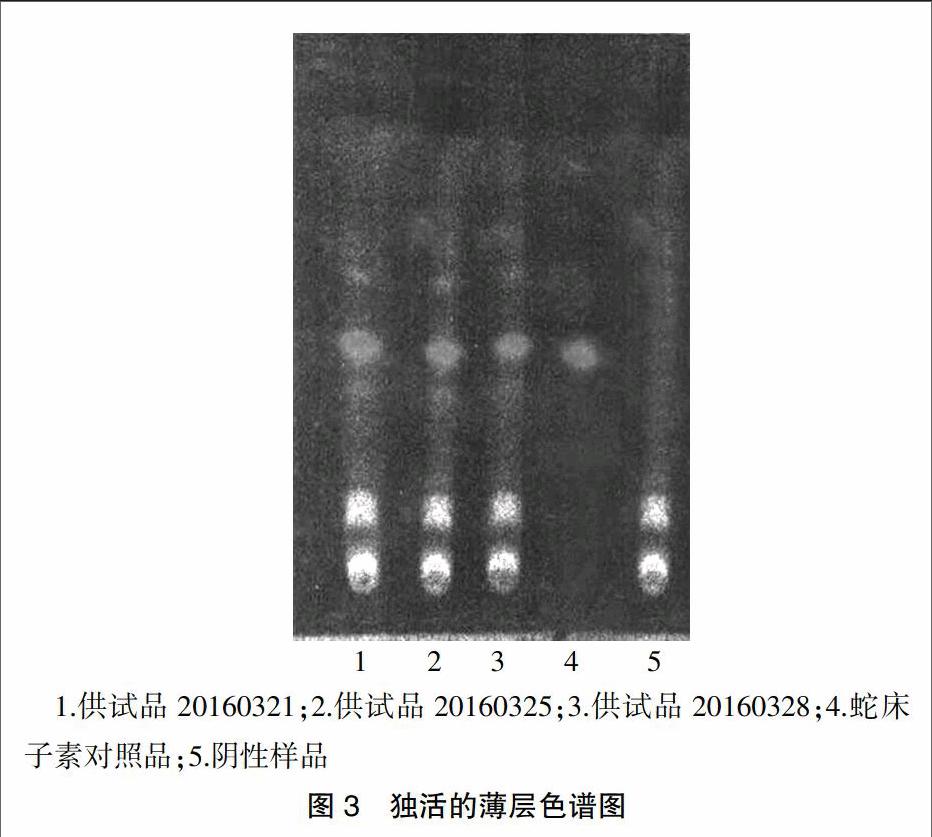
取本品内容物5 g,研细,加水30 ml使溶解,再用乙醚振摇提取3次,每次25 ml,合并上层乙醚液,挥干,残渣加甲醇1 ml溶解作为供试品溶液。再取蛇床子素对照品,用甲醇溶解制成1 mg/ml的溶液,作为对照品溶液。为增加鉴别的专属性,取去除独活的模拟处方制成阴性样品,再按上述供试品溶液的制备方法依法制成缺独活的阴性样品溶液。按照薄层色谱法《中国药典》2015年版一部附录Ⅵ B试验,分别吸取供试品溶液、阴性样品溶液各15~20 μl以及对照品溶液5 μl,点于同一硅胶G薄层板上,正己烷-醋酸乙酯(9∶3)作为展开剂,展开后取出晾干,置于紫外光灯(365 nm)下检视。结果显示,供试品色谱与对照品色谱在相应的位置上显相同颜色的斑点(图3)。
2.4狗脊的薄层鉴别
取本品内容物5 g,加水30 ml使溶解,加盐酸调节pH值至2,用乙醚振摇提取3次,每次25 ml,合并上层乙醚液,蒸干,残渣用1 ml甲醇溶解作为供试品溶液。再取原儿茶酸对照品,加甲醇溶解制成1 mg/ml的溶液作为对照品溶液。为增加鉴别的专属性,取去除狗脊的模拟处方制成阴性样品,再按上述供试品溶液的制备方法依法制成缺狗脊的阴性样品溶液。按照薄层色谱法《中国药典》2015年版一部附录Ⅵ B试验,分别吸取供试品溶液、阴性样品溶液各15~20 μl以及对照品溶液5 μl,点于同一硅胶G薄层板上,展开剂为三氯甲烷-甲醇-甲酸(6.0∶1.0∶0.2),展开后取出晾干,再喷5%三氯化铁乙醇溶液,于105℃加热至斑点清晰显色。结果显示,供试品色谱与对照品色谱在相应的位置显相同颜色的斑点(图4)。
2.5色谱条件与溶液制备
2.5.1色谱条件 色谱柱:Inertsil ODS-3反相色谱柱(5 μm,250 mm×4.6 mm);流动相:乙腈-0.1%磷酸溶液(28∶72);流速:1 ml/min;柱温:35℃;检测波长:270 nm;进样量:20 μl。理论板数以淫羊藿苷色谱峰计应≤1000。在此条件下,分别得到对照品和样品的分离色谱图(图5)。
2.5.2对照品溶液的制备 精密称取适量淫羊藿苷对照品,置于100 ml容量瓶中,加甲醇溶解制成浓度为52.5 μg/ml的溶液,即得。
2.5.3供试品溶液的制备 取装量差异下的本品内容物,研细,精密称定2.0 g,置具塞锥形瓶中,精密移取70%乙醇25 ml,称定重量,静置过夜,超声处理(功率250 W,频率35 kHz)30 min,冷却至室温,称定重量,再用甲醇补足减失的重量,摇匀滤过,取续滤液,即得[6-8]。
2.6方法学考察
2.6.1专属性试验 用固定阈值为990的方法来检测样品中淫羊藿苷的色谱峰纯度,得出峰的纯度因子为1000,在阈值之上,说明样品中的淫羊藿苷色谱峰为单一物质峰(图6)。
2.6.2线性关系考察 精密吸取淫羊藿苷对照品溶液4、12、20、28、40 μl,分别注入高效液相色谱仪,以进样量(0.21、0.63、1.05、1.47、2.10 μg)为横坐标,相应的峰面积为纵坐标,绘制标准曲线。测得线性回归方程为:Y=4×106X+192 488,r=0.998 4,线性范围为0.21~2.1 μg(表1)。
2.6.3精密度试验 取“2.5.3”项下的对照品溶液,按“2.5.1”项下的色谱条件连续进样6次,测定淫羊藿苷的峰面积。结果淫羊藿苷峰面积的RSD为0.68%(n=6),表明该方法的仪器精密度良好。
2.6.4稳定性试验 取“2.5.3”项下的供试品溶液,分别于0、2、4、6、8、12、24 h后进样,测定供试品溶液中淫羊藿苷色谱峰峰面积,计算RSD为0.25%(n=7),表明供试品溶液在制备后的24 h内基本稳定。
2.6.5重复性试验 取同一批腰痛康胶囊样品6份,每份约2.0 g,精密称定,按“2.5.3”项下的方法制备供试品溶液并按“2.5.1”项下的色谱条件测定含量。结果淫羊藿苷的平均含量为0.3700 mg/g,RSD为0.89%(n=6),表明该方法重复性良好。
2.6.6加样回收率试验 分别称取同一批次的腰痛康胶囊样品(淫羊藿苷含量为0.37 mg/g)5份,每份约1.0 g,精密称定,分别置于已加标并经氮气吹干的5个锥形瓶中(105 μg/ml的淫羊藿苷对照品溶液各5 ml),精密加入70%乙醇20 ml,再按“2.5.3”项下的方法制备;分别精密吸取20 μl,注入液相色谱仪,测定淫羊藿苷色谱峰面积,计算回收率,结果淫羊藿苷的平均回收率为99.2%,RSD为0.25%(n=5),表明该方法的准确性良好(表2)。
2.7样品测定
取7批腰痛康胶囊样品,分别按“2.5.3”项下方法制备供试品溶液,并按“2.5.1”项色谱条件进行测定,每批样品各测3次,以外标法计算各指标成分含量,(表3)。
3讨论
本文考察了不同浓度的乙醇或甲醇溶液与不同的超声提取时间对淫羊藿苷的提取效果的影响,结果表明70%的乙醇超声30 min的效果较为合适,提取充分且效果稳定;此外本实验还考察了先用乙醚进行提取,继而弃去乙醚液,然后再采用乙酸乙酯提取,结果发现回收率明显偏低,且操作复杂。在色谱条件摸索过程中,大量研究采用乙腈-水(30∶70)作为流动相,但试验所得的对照品色谱峰有拖尾现象,故考虑加酸以改善其峰形[9-11]。因此,选择常用的0.1%磷酸水溶液替代纯水溶液[12-13],结果发现对照品峰形有所改善。文献报道,淫羊藿苷检测波长为270 nm[14-17]。本试验采用UV-2550紫外分光光度计,在200~400 nm波长范围内对淫羊藿苷对照品溶液进行光谱扫描,测得在270 nm处有最大吸收,与文献报道相同。
腰痛康胶囊作为南昌市洪都中医院协定处方制剂,在我院临床应用范围广,治愈率高,对于急慢性腰痛、腰腿疼痛等有显著的疗效。因此,建立腰痛康胶囊的质量标准具有重要的临床价值。上述实验结果表明,本试验所建立的方法可操作性强,简便易行,同时专属性较强,具有较好的重现性,方法稳定且回收率高,可为腰痛康胶囊的质量控制提供可靠的依据。
[参考文献]
[1]孙清廉.补肾壮阳良药淫羊藿[J].家庭中医药,2013,(1):48-49.
[2]朱鸿飞,郑军,徐小燕,等.不同浓度淫羊藿苷修复骨损伤:争议与探索[J].中国组织工程研究,2014,18(2):301-306.
[3]尹志峰,刘敬闪,张兰桐.淫羊藿苷的含量测定方法[J].中国医院药学杂志,2003,23(8):493-495.
[4]李健英,王连芝.壮骨灵胶囊中淫羊藿苷的含量测定[J].中医药信息,2011,28(6):108-110.
[5]王宝全,赵庆华,刘忠良,等.不同厂家安神补脑液中淫羊藿苷含量考察[J].实用医药杂志,2011,28(2):140-141.
[6]宋霞,宋三孔,焦海胜.HPLC法测定乳病消片中淫羊藿苷含量[J].中国药师,2011,14(2):282-284.
[7]贾丹兵,刘珊,李乃民,等.HPLC测定参姜锁阳益气片中淫羊藿苷的含量[J].中国实验方剂学杂志,2011,17(1):35-36.
[8]国家药典委员会.中国药典[S].一部.北京:中国医药科技出版社,2015:1095-1096.
[9]尹志峰,刘敬闪,张兰桐.淫羊藿苷的含量测定方法[J].中国医院药学杂志,2003,23(8):493-495.
[10]张爱丽,齐凤琴.HPLC法测定补肾健骨片中淫羊藿苷的含量[J].中国药师,2010,13(11):1676-1677.
[11]崔俊凤,傅勇,耿海霞.RP-HPLC法测定益肾灵颗粒中淫羊藿苷的含量[J].中国药事,2005,19(9):546-547.
[12]黄耀广,陈秀英.RP-HPLC法测定回春胶囊中淫羊藿苷的含量[J].中国药事,2008,22(1):52-53.
[13]李苓.HPLC测定力多口服液中淫羊藿苷的含量[J].中国中医药信息杂志,2003,10(9):33-33.
[14]涂兴明,李赐恩,吴康郁.HPLC法测定骨康口服液中淫羊藿苷的含量[J].中药新药与临床药理,2013,24(6):613-615.
[15]俞吉,朱裕林,桑冉,等.高效液相色谱法测定骨疏灵颗粒中淫羊藿苷含量[J].实用药物与临床,2015,18(6):691-693.
[16]施元洪,刘镇峰,李镇波,等.高效液相色谱法测定复方清肺饮颗粒中淫羊藿苷的含量研究[J].中国当代医药,2015,22(12):7-9.
[17]张薇,鄂立勋,阚鹏甲.HPLC法测定补肾强身片中淫羊藿苷含量[J].中国当代医药,2009,16(25):38-38.
