玉竹中4种高异黄烷酮的二次层析薄层色谱法快速鉴别研究*
2020-04-11瞿志杰李美丽
张 颖,白 晶,吕 振,瞿志杰,李美丽,徐 涛,李 莉△
(1.哈尔滨商业大学药学院·药物工程技术研究中心,黑龙江 哈尔滨150076;2.齐齐哈尔医学院药学院,黑龙江 齐齐哈尔161006)
玉竹是百合科植物玉竹Polygonatum odoratum(Mill.)Druce的干燥根茎,药食同源[1],富含20余种高异黄酮成分[2-11],其中4种高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ含量相对较高,且具有较强的生物活性。高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ均具有较强的抑菌活性[12],高异黄烷酮Ⅰ,Ⅲ,Ⅳ对蛋白质非酶糖基化半数抑制量(IC50)分别为107.10,46.05,56.30 μmol/L[13],高异黄烷酮Ⅲ,Ⅳ对人白血病细胞(K562)、人肺癌细胞(A549)、人结肠癌细胞(HCT-15)的IC50分别为6.5,20.8,8.5 μg/mL和26.2,25.2,15.3 μg/mL[14]。2015年版《中国药典(一部)》中玉竹中药效物质的检测方法仅有多糖总量不低于6.0%与醇提物总量不低于50.0%,未对高异黄烷酮成分进行质量控制[15],不能全面反映玉竹质量。本研究中建立了玉竹中4种高异黄烷酮活性成分的薄层色谱(TLC)鉴别方法,旨在进一步控制其质量。现报道如下。
1 仪器与试药
1.1 仪器
C18-H固相萃取柱(4 000 mg/20 mL,BESEP,Germany);GF254铝制薄层板(10 cm×10 cm,10 cm×11 cm,Merck KGaA Darmstadt,Germany);XS3DU型电子分析天平(Mettler Toledo公司,精度为0.000 001 g);SC-5型紫外分析仪(北京金剑之光医药信息技术中心);双槽层析缸(120 mm×115 mm×5 mm,上海鼎杰生物科技有限公司);N-1001型旋转蒸发仪(上海爱朗仪器有限公司);微量进样器(上海高鸽工贸有限公司,10 μL);定量毛细管(杭州驰成医药科技有限公司,10 μL)。
1.2 试药
二氯甲烷、甲醇、甲酸、乙醇、石油醚、乙酸乙酯均为市售分析纯;高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ对照品[自制,经高效液相色谱-质谱(HPLC-MS)法和核磁共振波谱法(NMR)鉴定,纯度均大于98.0%];20批玉竹(按2015年版《中国药典(一部)》检验均符合规定)来源见表1,其中编号1~15为饮片、16~20为药材。
表1 不同批次玉竹的基本信息
2 方法与结果
2.1 溶液制备
对照品溶液:分别取4种高异黄烷酮对照品Ⅰ,Ⅲ,Ⅳ,Ⅴ适量,精密称定,分别用甲醇配制成0.4 mg/mL的溶液,置棕色瓶中,密闭,即得。
供试品溶液:取干燥玉竹粗粉约3.3 g,精密称定,置圆底烧瓶中,加乙醇14 mL,浸泡15 h,水浴(80℃)加热回流2 h,取上清液,如上重复提取1次,合并2次上清液,趁热抽滤,将滤液转移至蒸发皿中,水浴(78℃)蒸干,加入7 mL水,振摇,得浑浊液,转移至C18-H固相萃取柱中,将水溶液滤净,再用7 mL水洗脱,弃去水溶液;用10 mL甲醇缓缓洗脱上述C18-H固相萃取柱,将洗脱液置蒸发皿中,水浴(78℃)蒸至约1 mL,转移至1.5 mL棕色瓶中,用氮气吹干,再用100 μL甲醇定容,密闭。
阳性样品溶液:以玉竹S1和S6[采用超高效液相色谱(UPLC)法检测,高异黄烷酮Ⅲ及Ⅳ的含量大于40 μg/g,高异黄烷酮Ⅰ及Ⅴ的含量大于6 μg/g]作为阳性样品或对照药材。分别取其粗粉约3.3 g,按供试品溶液制备方法制得。
阴性样品溶液:以玉竹S16(采用UPLC检测,高异黄烷酮Ⅲ及Ⅳ的含量低于3 μg,高异黄烷酮Ⅰ及Ⅴ的含量低于0.6 μg/g)作为阴性样品。取其粗粉约3.3 g,按供试品溶液制备方法制得。
模拟阳性样品溶液:取阴性样品粗粉约3.3 g,精密称定,置圆底烧瓶中,加入高异黄烷酮Ⅲ90 μg,高异黄烷酮Ⅳ80 μg,高异黄烷酮Ⅴ24 μg,高异黄烷酮Ⅰ8 μg,作为模拟阳性样品。按供试品溶液制备方法制得。
2.2 薄层色谱法建立
2.2.1 色谱条件选择
4种高异黄烷酮化学结构(图1)相近,极性差异小,主要区别在于8位与4′取代基是甲基、甲氧基还是羟基,故在同一色谱条件下,很难将4个化合物有效分离。

图14 种高异黄烷酮化学结构式
采用GF254薄层板,以三氯化铁试液显色,分别考察了甲苯-乙酸乙酯-甲酸、甲苯-甲醇-甲酸、石油醚-甲醇、石油醚-乙酸乙酯、石油醚-乙酸乙酯-甲酸5个非极性及弱极性展开系统的TLC。结果显示,石油醚-乙酸乙酯(3∶2,V/V)对4种高异黄烷酮的分离效果最佳,但无法有效分离高异黄烷酮Ⅳ及Ⅴ。详见图2A。
采用GF254薄层板,以254 nm波长检视,分别考察了二氯甲烷-甲醇、二氯甲烷-甲醇-甲酸、二氯甲烷-甲醇-水3个中等极性展开系统的TLC。结果显示,二氯甲烷-甲醇-甲酸(150∶5∶3,V/V/V)对4种高异黄烷酮的分离效果最佳,但无法有效分离高异黄烷酮Ⅲ及Ⅳ。见图2 B。
2.2.2 二次层析薄层色谱法
分别取4种高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ对照品溶液、对照药材与供试品溶液各10 μL,点于同一GF254薄层板上,置含有10 mL二氯甲烷-甲醇-甲酸(150∶5∶3,V/V/V)的层析缸中,饱和,层析,取出,晾干,于254 nm或365 nm波长处检视与定位(通常高异黄烷酮Ⅲ,Ⅳ不能有效分离),在对照品高异黄烷酮Ⅴ与高异黄烷酮Ⅳ间画一条平行于底边的直线,沿此线将该TLC板剪裁成两部分。立即将下半部分用三氯化铁试液喷雾,并用滤纸将水吸干,对照药材、供试品溶液主斑点的位置及颜色均应与高异黄烷酮Ⅴ对照品主斑点相同;将上半部分用6 mL石油醚-乙酸乙酯(3∶2,V/V)二次层析,立即用三氯化铁试液检视,对照药材、供试样品主斑点的位置及颜色均应分别与高异黄烷酮Ⅰ,Ⅲ,Ⅳ对照品主斑点相同。
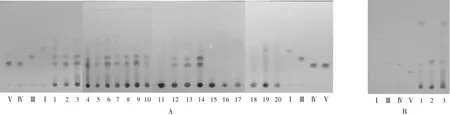
图2 薄层色谱图
2.3 方法学验证
2.3.1 阴性样品干扰试验
分别取4种高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ对照品、阳性样品、模拟阳性样品与阴性样品溶液,各10 μL,点于同一GF254薄层板上,按2.2.2项下方法操作。结果见图3。阳性样品、模拟阳性样品主斑点的位置、颜色分别与4种对照品的主斑点相同,而阴性样品无相应斑点,说明玉竹中其他成分对高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ检测结果无干扰,方法有较强的专属性。
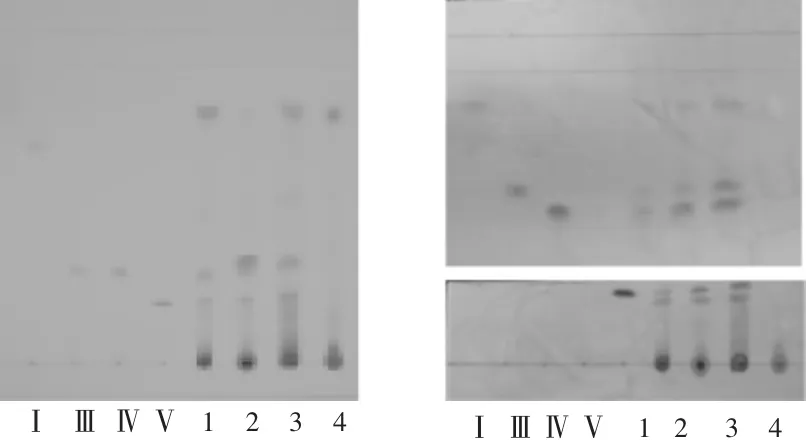
图3 阴性样品对4种高异黄烷酮薄层色谱的干扰试验
2.3.2 检测限(LOD)考察
分别量取0.7,1.3,2.5,5.0,10.0 μL高异黄烷酮Ⅰ,Ⅳ,Ⅲ,Ⅴ对照品溶液(相当于高异黄烷酮0.3,0.5,1.0,2.0,4.0 μg),依次点于同一块GF254薄层板上,按2.2.2项下方法操作,高异黄烷酮Ⅰ,Ⅲ,Ⅳ的展开剂为二氯甲烷-甲醇-甲酸(150∶5∶3,V/V/V),高异黄烷酮Ⅴ的展开剂为石油醚-乙酸乙酯(3∶2,V/V),依次在254nm和365 nm波长处与三氯化铁试液显色项下检视,结果见表2与图4。三氯化铁试液检视的灵敏度较高,高异黄烷酮Ⅰ,Ⅳ,Ⅲ,Ⅴ的LOD分别为2.0,2.0,1.0,1.0 μg。
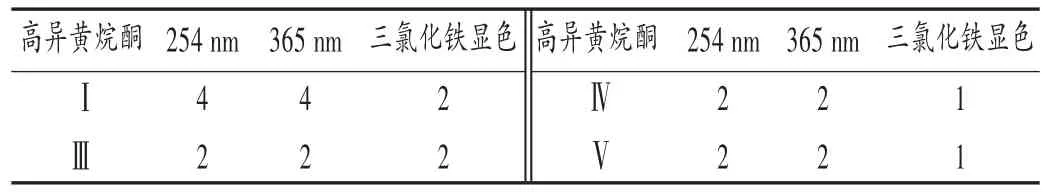
表2 不同检视方法下4种高异黄烷酮TLC的LOD(μg)

图44 种高异黄烷酮薄层色谱的检测限
2.3.3 稳定性考察
供试品溶液:分别取4种高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ对照品与供试品溶液适量,置1.5 mL加盖棕色瓶中,放置0,1,2,4,8,24 h后,按2.2.2项下方法操作,依次在254 nm和365 nm波长处与三氯化铁试液显色项下检视,结果6个时间点的TLC主斑点无显著差异,表明供试品溶液在24 h内稳定性较好。
TLC三氯化铁试液显色:按2.2.2项下方法操作,分别取4种高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ对照品溶液,点于同一GF254薄层板上,在365 nm波长下检视。在室温下,1 h后Ⅲ,Ⅴ主斑点的颜色与面积逐渐变淡、变小,高异黄烷酮Ⅰ,Ⅳ24 h内未变,详见图5 A;在加热(80℃)条件下,10 min后高异黄烷酮Ⅲ,Ⅴ主斑点的颜色与面积逐渐变淡、变小,高异黄烷酮Ⅰ,Ⅳ60 min内未变,详见图5 B。可见,高异黄烷酮Ⅲ,Ⅴ在空气中的稳定性较差,且加热可加速其氧化变质。按2.2.2项下方法操作,将4种高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ对照品和供试品溶液的TLC用三氯化铁显色后,观察室温下0,1,2,4,8,24h时的变化,24 h内主斑点的颜色与大小无显著差异,表明显色后的成分较稳定。
可见,4种高异黄烷酮溶液及其TLC三氯化铁试液显色后均较稳定,但TLC主斑点显色前不稳定,特别是高异黄烷酮Ⅲ,Ⅴ在加热条件下极不稳定,可能源于高异黄烷酮Ⅲ,Ⅴ结构中8位含有游离的酚羟基(见图1),在空气中极易被氧化。建议2.2.2项下层析后采用自然晾干,并立即用三氯化铁试液显色,可有效避免假阴性现象。

图5 不同条件下4种高异黄烷酮薄层色谱图(365 nm)
2.4 样品检测
将4种高异黄烷酮对照品Ⅰ,Ⅲ,Ⅳ,Ⅴ和20批玉竹样品(S6为对照药材)溶液按序号依次点于同一GF254薄层板上,按2.2.2项下方法操作,结果见图6与表3。可见,有5批玉竹(S1~S4,S6)检出高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ,8批玉竹(S5,S7~S10,S12~S14)仅检出高异黄烷酮Ⅲ,Ⅳ,7批玉竹(S11,S15~S20)未检出高异黄烷酮。
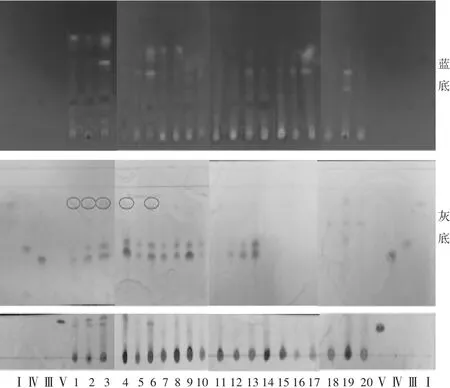
图6 二次层析薄层色谱法鉴别玉竹中4种高异黄烷酮(蓝底-365 nm;灰底-三氯化铁显色)

表320 批玉竹中高异黄烷酮检测结果

图720 批玉竹中4种高异黄烷酮的检出情况
2.5 不同产地玉竹质量差异
按表3将玉竹中高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ检测结果进行归类分析,结果见图7。可见,20批玉竹中检出高异黄烷酮Ⅲ,Ⅳ的共13批,其中5批均检出高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ,8批检出高异黄烷酮Ⅲ,Ⅳ。此外,还有7批玉竹中均未检出高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ。说明不同产地玉竹中高异黄烷酮存在显著差异。
3 讨论
3.1 样品提取条件选择
取同一批玉竹,按2.1.2项下方法操作,分别将不同条件下获得的玉竹提取液用UPLC及TLC检测。结果,分别用粗粉与碎块2种形态进行提取,前者提取效率较高;分别以70%乙醇、80%乙醇、95%乙醇、无水乙醇、甲醇作为提取溶剂[16],除前2种提取效率较低外,其他提取效率相近,因95%乙醇无害、价廉,故以此作为本研究中的提取溶剂;分别采用超声与回流提取,后者提取效率更高[17];分别采用水沉后离心过滤与水沉后用C18-H固相萃取柱处理作为纯化手段,后者TLC对主斑点干扰小。
3.2 薄层色谱展开方式选择
由4种高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ的TLC图谱可见,在石油醚-乙酸乙酯(3∶2,V/V)中高异黄烷酮Ⅳ,Ⅴ未分离,仅能鉴别高异黄烷酮Ⅰ,Ⅲ;而在二氯甲烷-甲醇-甲酸(150∶5∶3,V/V/V)中,Ⅲ,Ⅳ不能分离,仅能鉴别高异黄烷酮Ⅰ,Ⅴ。由2.2.2项下可见,通过将上述2种展开方式整合,剪掉首次层析后TLC中Rf值最低的主斑点(Ⅴ),对混合物斑点(Ⅲ,Ⅳ)进行二次层析,可实现在同一薄层板上有效分离4种高异黄烷酮成分。
3.3 不同产地玉竹质量分析
20批玉竹中有7批未检出高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ。根据TLC点样量、LOD及供试品溶液取样量进行推算,每1 g玉竹中高异黄烷酮Ⅰ及Ⅲ的含量低于6 μg,高异黄烷酮Ⅳ及Ⅴ的含量低于3 μg时,即不能用本研究中的方法检出。故该方法实质是对4种高异黄烷酮进行限量控制的基本方法,可对不同产地玉竹质量进行初步评价。20批玉竹中,13批检出高异黄烷酮Ⅲ及Ⅳ。该成分不仅具有抗癌活性,且在TLC中斑点颜色明显深于高异黄烷酮Ⅰ及Ⅴ;经UPLC验证,高异黄烷酮Ⅲ及Ⅳ的含量是高异黄烷酮Ⅰ及Ⅴ的3~5倍,现有玉竹质量标准可增设Ⅰ,Ⅲ,Ⅳ,Ⅴ的TLC鉴别与Ⅲ及Ⅳ的含量测定方法。
综上所述,本研究中依据2015年版《中国药典(一部)》,分别对20批市售玉竹进行了检验,其灵敏度、专属性强,结果均符合规定,但7批未检出高异黄烷酮Ⅰ,Ⅲ,Ⅳ,Ⅴ的玉竹中多糖含量明显高于13批检出高异黄烷酮Ⅲ及Ⅳ的玉竹。这种成分差异很可能会导致不同产地玉竹的疗效与保健功能的不同,希望能引起关注,进一步研究不同产地玉竹药理作用与保健功能的异同。
