河南一腓骨肌萎缩症家系分析并文献复习
2019-12-25孙瑞华时英英夏明荣张杰文
孙瑞华 时英英 夏明荣 张杰文
河南大学人民医院、河南省人民医院神经内科,河南 郑州 450003
腓骨肌萎缩症(peroneal muscular atrophy)作为一种最常见的外周神经退行性遗传病,发病率约1/2500[1],又称为夏科一马利一杜斯氏病(Charcot-Marie-Tooth disease,CMT)[2]。此病出现感觉神经及运动神经受累,临床表现为进行性远端感觉减退、肌无力和肌萎缩,又称为遗传性运动感觉性神经病(heriditary mater and sensory neuropathy, HMSN)。CMT有常染色体显性遗传、常染色体隐性遗传、X连锁显性和隐性遗传四种遗传方式。根据遗传方式和神经病变的不同,将CMT分为脱髓鞘型CMT(也称CMT1),轴突型CMT(也称CMT2),中间型CMT(也称DI-CMT)。
1 资料和方法
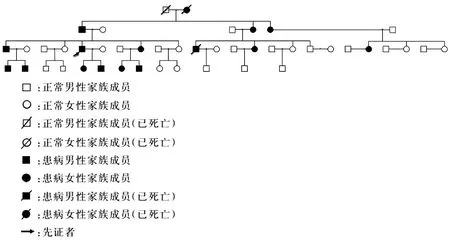
1.1临床资料患者男性,54岁,河南人,高中学历。以“进行性双足畸形、双下肢麻木无力40年。”为主诉于2018-03-21入院。患者自幼双足呈“马蹄足畸形”,同时双脚并拢时无法蹲下,须双脚分开扶持才能蹲下,弹跳力差,近几年来逐渐出现四肢肌肉萎缩,以双侧小腿、手掌明显,行走时稍有困难,曾多次发生跌倒,半月来出现双下肢感觉减退,双足袜套样改变,否认视物成双、进食呛咳、听力下降等症状,既往有2型糖尿病病史5 a,口服格列喹酮(30 mg bid po)、二甲双胍缓释片(0.5g bid po),血糖控制一般。8 a前跌倒出现右侧胫腓骨骨折,于当地医院行内固定,目前钢板已经取出,否认高血压、脑血管疾病病史,否认肝炎、结核、疟疾等传染史,否认输血、献血史,否认食物及药物过敏史,预防接种随当地社会进行。家族史:四代中,奶奶、父亲有腓骨肌萎缩及马蹄足畸形,大姑、二姑有马蹄足畸形;有1哥1妹,均有马蹄足畸形;1表哥2表妹有马蹄足畸形;有1女,有马蹄足畸形,1子双下肢感觉减退;2侄子有马蹄足畸形,1外甥、1外甥女双下肢感觉减退。总结来讲,患者四代中,共45人,有症状者有16人,其中男性8人,女性8人,3人同时有腓骨肌萎缩、马蹄足及双下肢感觉减退,10人有马蹄足及双下肢感觉减退,3人无腓骨肌萎缩及马蹄足,仅伴有双下肢感觉减退,仅先证者一人患有糖尿病,症状最重。查体:四肢肌张力正常,肌力:左上肢Ⅴ级,左下肢Ⅴ级,左下肢Ⅴ级,右下肢Ⅴ级,双足背屈肌力Ⅱ级。肱二头肌反射、肱三头肌反射、桡骨膜反射、膝腱反射、跟腱反射均消失,踝阵挛阴性。病理反射阴性、脑膜刺激征阴性、共济运动正常。感觉系统检查,双踝关节以下浅感觉减退。
辅助检查:抽血检查:血常规、免疫全套、 肝功、肾功、电解质、血沉、CC-反应蛋白、尿常规、病毒、甲功、叶酸、维生素B12、肿瘤标记物、粪常规、心肌酶、凝血等检查未见明显异常。副肿瘤抗体:抗-Amphiphysin、抗-CV2、抗-PNMA2、抗-Ri、抗-Yo、抗-Hu均为阴性。葡萄糖 7.38 ↑ 3.88~6.11 mmol/L;糖化血红蛋白5.9%。彩超检查示:左室松弛功能减退;甲状腺左侧叶实性结节;左侧颈总动脉斑块形成;脂肪肝;肝囊肿;右肾结石;右侧股总动脉斑块形成;双侧股浅、股深、腘、胫前、胫后、足背动脉内膜钙化;双侧足背动脉流速减低;双侧胫前动脉部分管腔血流充盈差(考虑局部狭窄)。磁共振检查示:轻微脑白质脱髓鞘;磁敏感加权成像(Susceptibility weighted imaging,SWI)未见明显异常;脑磁共振血管成像(Magnetic resonance angiography,MRA)未见明显异常;左侧上颌窦囊肿,双侧筛窦、上颌窦轻度炎症;左侧下鼻甲肥大,鼻中隔稍右偏。心电图:窦性心律;逆钟向转位;部分导联ST-T异常。骨密度检查报告示:腰椎1~4:骨量正常;左侧股骨部位:低骨量(参考标准:骨量正常:T值≥-1.0SD;低骨量:2.5SD 图1 A、B、C分别表示先证者、先证者哥哥、先证者女儿PMP22 基因外显子及其附近区域存在大片段重复Figure 1 A,B,and C respectively represent PMP22 gene exon and its vicinity in proband,proband's brother and proband's daughter exist large fragment repeat 图2 家系图Figure 2 Family diagram 1.2研究方法 1.2.1 电生理检查:经患者及家属同意,在河南省人民医院神经电生理检查室对患者进行四肢电生理检查。其他家属家族成员已故或不能出现或者拒绝接受有创检查,遂未行电生理检查。 1.2.2 基因检测:经患者及家属同意,将患者及其2名亲属(女儿、哥哥)周围血样于送至北京信诺佰世医学检验所,采用多重连接探针扩增技术(MLPA)(P033) 检测待检样本基因外显子缺失/重复变异,并以正常DNA作对照,数据采用Coffalyser软件分析进行基因检测。其他家族成员已故或不能出现,遂未送检样品。 2.1电生理检查双正中、尺神经运动传导波幅降低、潜伏期明显延长、速度明显减慢,无表现离散及传导阻滞,感觉传导未见波形,左正中神经F波潜伏期明显延长;双胫后神经、腓总神经运动传导无波,腓肠神经感觉传导无波;双第一骨间肌、胫前肌见自发电位及高宽运动单位电位(MUP),胫前肌主动收缩募集减少;左股直肌未见自发电位,主动收缩募集正常。结论:四肢多发性周围神经病,脱髓鞘继发轴索损害,患者的电生理表现、临床表现结合家族史考虑:CMT1型。 2.2基因检测先证者PMP22 基因外显子及其附近区域存在大片段重复;经 MLPA 验证,先证者哥哥和先证者女儿 PMP22 基因外显子及其附近区域均存在大片段重复(图1)。由此确诊病人及家属为常染色体显性遗传的CMTI型,家系图见图2。 腓骨肌萎缩症为常见的遗传性周围神经病,大多数患者于儿童时期发病,少数在成年发病,病程进展缓慢[3]。儿童和青年晚期发病,呈慢性进行性腓骨肌萎缩,有遗传倾向等特征,是遗传性周围神经病中最常见的类型。 3.1临床表现大多数CMT患者先累及双侧下肢,且受累程度较一致,少数患者可出现一侧先起病,一侧肌萎缩较明显。CMT的典型临床表现为:对称性双下肢远端肌无力和(或)肌萎缩,可呈“鹤腿”样改变,双上肢受累时可出现爪形手,下肢重于上肢。但不影响寿命。 3.2发病机制CMT发病机制不明,现已发现多种CMT致病基因,这些致病基因可从多个方面发挥作用。目前,已发现超过80种基因的突变导致多种多样形式的CMT和远端遗传性运动神经病变,这对患者、家属和临床医生具有挑战性[4]。在西方国家,80%以上的CMT由PMP22、MPZ、MFN2和缝隙连接蛋白β1(gap junction protein beta 1,GJB1)基因突变导致[5]。据报道,大多数的CMT1患者由17p11.2-12区的1.5 Mb[6]的正向串联重复突变所致。LupskiLUPSKI等[7]研究6个CMT1A大家系时首次发现PMP22基因突变,PMP22的基因位于重复区域并被鉴定为负责的疾病基因[8]。过度表达的PMP22可使髓鞘不稳定性增加,从而发挥作用。 3.3分型依据病理和电生理特点可分为两型:CMT1型,神经转导速度明显减慢,神经活检可见广泛的节段性脱髓鞘和髓鞘增生;CMT2型,其神经传导速度减慢不如CMT1型明显,神经活检可见轴突变性,脱髓鞘改变极少见。神经传导速度的测定是CMT分型最可靠而易行的手段,尤其是正中神经传导速度的测定。 3.4诊断标准CMT诊断标准[9]:(1)慢性起病,缓慢进展的肢体远端尤其是双下肢远端的肌无力和肌萎缩,典型者成鹤腿样畸形;(2)肌电图示神经源性受损;(3)神经活检证实为本病;(4)阳性家族史;(5)弓形足或脊柱侧弯;(6)排除其他病因。所有患者须具备上述第(1)、(2)、(6)3条加上第(3)、(4)、(5)中任何一条,或家族中已有确诊者,再具备(1)即可入选。 先证者慢性起病,幼年发病,为双下肢进行性对称性远端肌无力和肌萎缩,表现为典型的鹤腿样畸形,弓形足,伴有双上、下肢腱反射消失,伴有双下肢明显的感觉障碍,具有家族史,男女患病比例大致相等,神经电生理提示周围神经脱髓鞘样改变,基因检测患者、哥哥、女儿呈PMP22 基因外显子及其附近区域存在大片段重复,为典型的常染色体显性遗传的CMTI型。患者既往有糖尿病史,血糖控制一般,糖尿病的神经损害更加重了患者周围神经的损伤,较家族其他患病者来讲,先证者出现周围神经损伤的程度更深,症状更明显。之前的文献中也提到糖尿病患者在异常糖代谢途径中所造成的氧化应激损伤及微循环障碍,结合雪旺细胞(Sehwann cell)生长因子的分泌等造成糖尿病周围神经病变。以往报道了几例与糖尿病相关的CMT病例和家族[10-12]。一项回顾性研究评估了大量患有CMT1的患者,这些患者具有不同的合并症,包括糖尿病、肥胖、甲状腺机能减退和接触有毒物质。该研究报道了CMT1和糖尿病患者CMT1临床和神经生理学表现恶化的趋势[13]。本例患者未进行神经活检,因为其为有创检查,且现有证据已可确诊疾病。 3.5鉴别诊断临床上CMT1主要与慢性炎性脱髓鞘性多发性神经根神经病(CIDP)、遗传性压力敏感性周围神经病(HNPP)等相鉴别。 CIDP和CMTl二者临床表现相似,均有运动和感觉受累,两者鉴别比较困难。CIDP为自身免疫性疾病,病变为局灶性。CMT1为遗传病,神经受累程度相同。 PMP 22基因重复突变可引起CMT1,而PMP 22大片段缺失可导致HNPP[15]。 3.6治疗措施现在来讲,CMT 无特殊治疗,主要有适度运动及对症支持治疗,其中对症支持治疗包括康复治疗、药物治疗、外科矫形手术等,目标是最大限度地恢复独立活动能力、提高生活质量,最大限度地减少残疾的发生与发展。 应该根据患者年龄、肌力失衡范围和程度、骨骼畸形类型和程度、患者的治疗期望值等因素制定具体的治疗方案。与其他神经肌肉疾病相反,建议在次最大工作水平上进行有氧运动[16]。最近的一项研究表明,家庭成员和看护人不相信康复对他们的亲属有效(由于他们观察到相对较小的益处),但CMT患者从康复中感知身心受益[17]。患者足底屈肌无力在维持直立位置方面也存在更大的困难[18],使用踝足矫形器可以改善患者的步态[19]。研究发现,孕酮受体拮抗剂可用于减少PMP22蛋白的聚集,改善症状[20];维生素C可减少脱髓鞘,利于恢复肌肉功能[21];神经营养因子3利于轴索的修复。此外, 应该高度重视患者的心理问题,多学科协作治疗会更有利于疾病的救治[22]。若患者合并有糖尿病等可引起周围神经病变的高危因素,需要积极控制血糖在合适范围,避免加重病情。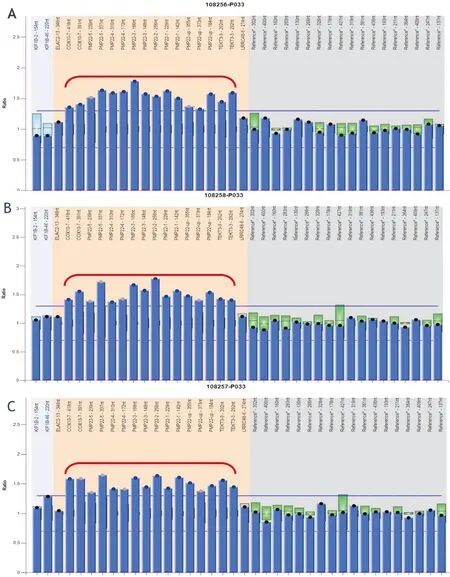
2 结果
3 讨论
