VCP基因突变致临床肌萎缩侧索硬化一例
2019-09-24王建徐祖才
王建 徐祖才
1 病例报告患者男,23岁。因“饮水呛咳、吞咽困难,四肢乏力2个月”于2018年2月入院。2个月前无明显诱因出现饮水呛咳、吞咽困难,只能进少许半流质饮食;讲话时吐词不清、言语含混;四肢乏力,以双上肢明显,表现为双手持物欠灵活、穿衣受限、双上肢不能平举,逐渐波及双下肢,出现步态不稳,上楼梯费力。曾就诊于当地医院,行颅脑CT检查显示小脑半球低密度影,颅脑MRI检查示左侧放射冠区腔隙性梗死,双侧小脑半球软化灶(未提供影像学图片)。给予对症治疗(方案不详)后患者症状无缓解,遂就诊于遵义医科大学附属医院。患者8岁时因外伤致双眼失明(具体不详),既往无特殊传染病及遗传家族史,无吸烟、饮酒史,起病以来体重下降约16 kg。入院查体:意识清楚,对答切题,言语含糊,反应力、定向力、记忆力、计算力正常,双眼球内陷,双眼失明,无光感,左侧鼻唇沟稍变浅,伸舌居中,咽反射减弱,颈软,四肢肌肉均有不同程度萎缩,双上肢肌力Ⅲ级,双下肢肌力Ⅳ级,四肢肌张力正常,右下肢膝反射、踝反射亢进,右侧Babinski征阳性,脑膜刺激征阴性。血常规、电解质、肝肾功能、心肌酶、肌钙蛋白、甲状腺功能、肿瘤标记物、凝血功能检查未见异常。脑脊液压力正常,脑脊液常规、生化指标均未见异常。肌电图(EMG)检查结果显示,四肢神经源性损害,以左下肢和右上肢明显,感觉系统正常。患者入院后给予改善微循环、营养神经、针灸、理疗及对症支持等治疗,症状仍逐渐加重。4 d后复查EMG结果示广泛脊髓前角运动神经元损害,脑干运动神经元损害可能;体感诱发电位检查示合并中枢体感通路受损病变;头颅CT检查示右侧小脑半球病变,左侧小脑半球局部脑沟增宽或软化灶可能;头颅MRI示双侧小脑半球软化灶,双侧大脑半球脱髓鞘病变,轻度脑萎缩改变(图1)。基因测序显示患者含缬酪肽蛋白(valosin-containing protein,VCP)基因异常,为c.266G>A(编码区第266号核苷酸由G变为A)的杂合核苷酸变异,该变异导致了第89号氨基酸由Arg变为Gln(p.Arg89Gln),为错义突变(图2)。患者父母及其弟弟均未见该位点异常变异。诊断:肌萎缩侧索硬化(ALS)。予以改善微循环、营养神经、针灸、理疗等治疗2周,症状无明显好转。
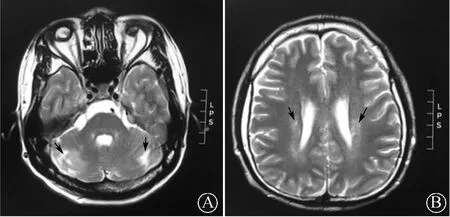
图1 患者头颅MRI检查结果双侧小脑长T2信号(A,箭头所示)、双侧半球多发斑片状长T2信号(B,箭头所示)
2 讨论ALS是一种以上、下运动神经元损害为特征的慢性、进行性神经变性疾病。临床主要表现为进行性的肌无力、肌萎缩,逐渐进展,感觉系统和括约肌功能一般不受累。该病于1869年被法国神经病学家Jean-Martin Charcot首次报道,临床罕见,年发病率约为(0.50~1.76)/10万,分为家族性和散发性。VCP基因突变是其病因之一。目前尚无有效治疗方法,患者平均生存期为3~5年。本例患者为青年男性,亚急性起病,临床表现为饮水呛咳、吞咽困难、四肢乏力,具有上、下运动神经元损害表现的体征,EMG检查示典型的神经源性损害,感觉系统未受累,头颅MRI提示双侧小脑半球软化灶,双侧大脑半球脱髓鞘病变,病灶主要分布在白质区域,考虑为运动神经损害可能,支持存在上运动神经元损害的证据,基因测序结果提示VCP错义突变,综合临床及实验室检查结果诊断为ALS。
遗传学研究发现了许多与ALS相关的遗传因素,这些因素最终可导致ALS运动神经元的退化[1]。VCP基因突变约占家族性ALS的1%~2%,散发性<1%。VCP是一种广泛表达的、具有多种细胞ATP酶活性的多功能成员,参与多个泛素依赖的细胞内过程[2]。研究表明,在VCP突变中,常染色体显性、杂合性错义变异与一组表型(也称为多系统蛋白病)有关,其中包括包涵体肌病(IBM)、早发型Paget病(PDB)、肌萎缩性脊髓侧索硬化14型伴或不伴额
颞叶痴呆(ALS-FTD),统称为IBMPFD[3]。有研究证实,VCP蛋白是泛素自噬体成熟的关键,而交互响应DNA结合蛋白-43(TDP-43)是泛素包涵体的主要组成部分,细胞内TDP-43蛋白的异常集聚会导致细胞毒性从而介导VCP的突变[4-5]。因此,目前认为VCP的错义突变会导致构象改变,最终导致细胞内弥漫性的泛素包涵体和TDP-43蛋白沉积。这可能是引起IBMPFD类疾病的发病机制。关于VCP基因突变可引起ALS早已被证实,而本例患者存在VCP基因c.266G>A位点突变,既往尚未见报道。该位点突变是否具有致病性,本研究小组拟进一步对该位点进行细胞功能实验验证。
