家族局部性脂质营养不良3型合并复发性胰腺炎一例临床分析及文献复习
2019-07-01吴润秋金玉杨辉
吴润秋 金玉 杨辉
南京医科大学附属儿童医院消化科,南京 210008
家族局部性脂质营养不良(familial partial lipodystrophy,FPLD)是由遗传、药物、炎症和自身免疫等原因导致的以机体脂肪组织不同程度缺失伴各种代谢紊乱为特征的疾病,是一种罕见的常染色体显性遗传病,人群中的发病率不到1/1 000万[1],目前全球报道约1000例。已知的FPLD相关基因共6种,包括LMNA、ZMPSTE24、PPARG、AKT2、CIDEC和PL1N1[2-3]。其中过氧化物酶体增殖物激活受体γ(PPARG)基因杂合突变(FPLD3型)是非常少见的类型,目前国外报道大约30例[2,4],国内未见报道[5]。FPLD3型临床表现为儿童、青少年或成年患者早期的对称部位脂肪缺失,以四肢、臀部和大腿部为主,残存的脂肪组织纤维化明显,但腹部皮下脂肪及内脏脂肪总量没有改变[6]。由于PPARG突变累及的相关通路在糖脂代谢过程中起重要作用,因此该型患者的糖脂代谢紊乱更加严重[7],可产生胰岛素抵抗、高脂血症、反复胰腺炎等并发症[8]。现报道1例临床和基因检测均明确诊断的FDLP3型患儿的临床特征及随访资料,并结合国内外文献进行分析,从而提高对该疾病的认识。
一、临床资料
患儿女,7岁5月,足月顺产,出生体重3.1 kg,出生时外貌正常。5岁3月时因“双侧肘、膝关节伸侧及臀部粟粒样脂肪瘤近1月”入院。家族史:患儿母亲4年前有“左肾上腺肿瘤”病史,父亲体健。实验室检查:三酰甘油明显增高,最高达26 mmol/L,行脂肪瘤切除手术后出院。患儿5岁10月时因“阵发性上腹痛伴呕吐3 d”第2次入院。实验室检查:空腹血糖7.92 mmol/L,血钙2.03 mmol/L,血淀粉酶2 430 IU/L,三酰甘油2.92 mmol/L,总胆固醇3.94 mmol/L,尿胰蛋白酶原Ⅱ弱阳性,尿淀粉酶3 119 U/L。腹部B超示胰腺增大,腹部CT示胰腺改变伴胰周渗出。入院后予空肠置管肠内营养,奥曲肽减少胰液分泌,奥美拉唑、头孢唑肟及甲硝唑抗感染治疗。5 d后复查腹部B超提示胰尾少许渗液;上腹部MRI平扫示胰腺体积增大,T2信号不均匀增宽,边缘模糊,呈长T1长T2信号,周围脂肪间隙模糊,可见渗出影;MRCP未见明显异常(图1);血淀粉酶 55 U/L,脂肪酶125 U/L,尿胰蛋白酶原Ⅱ阴性,尿淀粉酶97 U/L。11 d时再次复查腹部B超未见胰腺周围渗出而出院。患儿6岁4月时因“腹痛1 d伴呕吐数次”第3次住院。实验室检查:血钙2.31 mmol/L,三酰甘油9.54 mmol/L,总胆固醇4.25 mmol/L,血淀粉酶383 U/L,尿胰蛋白酶原Ⅱ阳性,尿淀粉酶2 047 U/L;MRI上腹部平扫+MRCP表现同第2次住院时。予胃肠减压、奥曲肽、奥美拉唑、头孢唑肟、蛇毒血凝酶1U(1次)及补液治疗后复查尿胰蛋白酶原Ⅱ阴性,尿淀粉酶89 U/L,血淀粉酶66 U/L而出院。患儿7岁5月时因“阵发性上腹痛1 d”第4次入院,无发热、呕吐、咳嗽及腹泻。腹部CT示胰腺肿胀伴周围渗出;实验室检查:三酰甘油73.26 mmol/L,总胆固醇14.32 mmol/L,血淀粉酶367 U/L。予胃肠减压、拉氧头孢、甲硝唑、奥美拉唑、善宁针、杜玛治疗,同时先予血浆置换再行血液灌流治疗,10 d后复查血总胆固醇6.36 mmol/L,高密度脂蛋白0.46 mmol/L,三酰甘油5.61 mmol/L,血淀粉酶46 U/L。
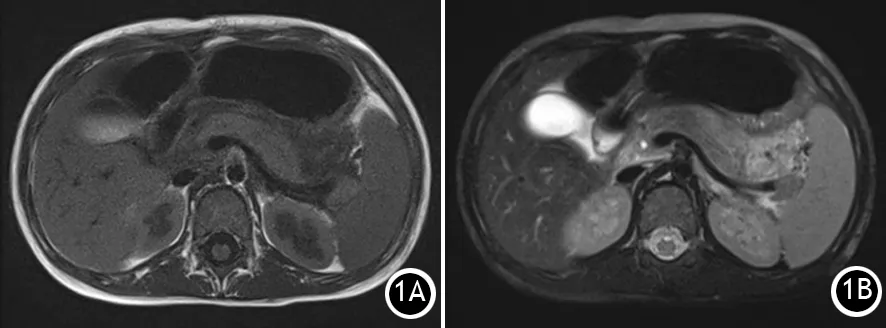
图1 患儿MRI平扫(1A)及MRCP(1B)征象
经家长知情同意,分别抽取患儿及父母外周血,采用靶向捕获-高通量测序进行糖、脂代谢疾病相关基因分析(北京德易临床检验所),结果采用基因检测智能操作系统进行注释,SNPEFF软件(hg19, GRCh37)进行变异解读,其中功能性的编码区和剪切位点的变异进入下一步分析。通过数据库查找变异位点或疑似致病突变的收录情况进行基因突变致病的注释,最终发现患儿PPARG基因第3外显子区域有1个已知杂合突变点,为c.70G>A,氨基酸突变为p.24,V>I,蛋白结构预测结果为有害。患儿父亲也发现同样的基因突变,但为正常临床表型。患儿母亲为野生型。提示患儿PPARG序列c.70G>A突变来源于父亲,导致氨基酸改变(图2)。
二、文献复习
以“家族局部性脂质营养不良3型”、“PPARG”为关键词检索中国知网、万方数据库及中国生物医学文献数据库相关中文文献,未检索到确诊的PPARG基因突变致FPLD3 型病例报道。以“familial partial lipodystrophy”、“PPARG”为关键词检索PubMed数据库,检索到30篇文献,排除非临床病例报道、临床资料不全及成年患者的文献,共2篇文献累计2例FPLD3型患儿进入汇总分析,2例患儿均无合并胰腺炎报道。
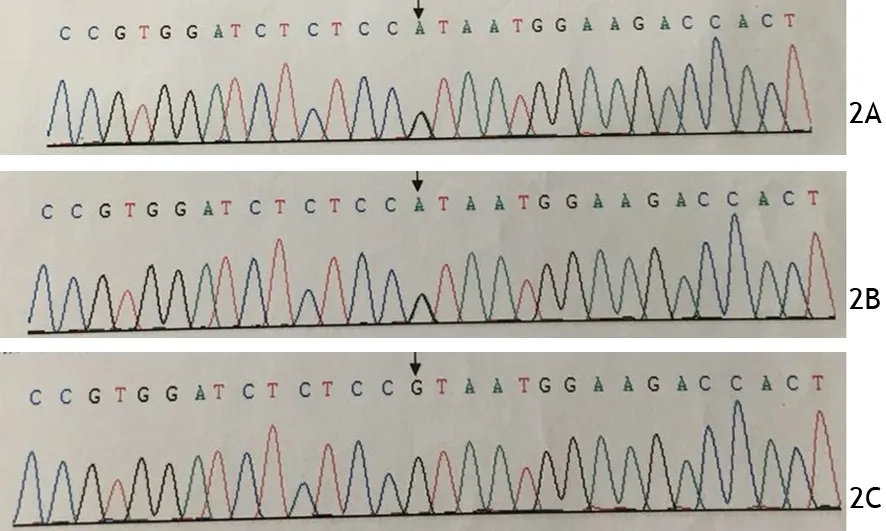
图2 患儿(2A)及其父亲(2B)、母亲(2C)的PPARG序列
文献复习的2例FPLD3型患儿均为女性,但种族背景各异,发病年龄分别为12岁和5岁,基因诊断年龄分别为14岁和12岁,诊断时间延迟多与基因检测技术限制以及既往对于该病认识有限有关。FPLD3型最常见的临床表现是四肢和臀部的皮下脂肪组织损失以及面部、颈部和躯干脂肪储存增多,但腹部皮下脂肪及内脏脂肪总量没有改变。1例患儿有该典型表现,其他临床表现和体征为高血压,黑棘皮征,心脏病变,月经周期不规律(表1)。实验室检查以高三酰甘油血症最为常见,其他异常包括肝脂肪变性(1例),胰岛素抵抗(1例)。2例患儿均无胰腺炎反复发作病史。

表1 2例FPLD3型患儿临床及PPARG基因特点
讨 论
脂质营养不良分为遗传性和获得性,遗传性脂质营养不良分为先天性全身脂质营养不良(congenital generalized lipodystrophy,CGL)和FPLD[11]。FPLD有5种亚型,为FPLD1~FPLD5[12],其遗传方式多样,可表现为常染色体显性遗传、隐性遗传或者是伴性遗传。FPLD2型是最常见也是最先报道的亚型,但目前也仅有报道约300例[11],FDLP3型与PPARG基因杂合突变有关。本例患儿5岁时双侧肘关节、膝关节伸侧及臀部出现粟粒样脂肪瘤,入院体检无特殊面容,无黑棘皮征,实验室检查示三酰甘油及空腹血糖明显升高,病程中反复出现3次胰腺炎,FPLD相关基因检测显示患儿PPARG 基因第3外显子存在1处杂合突变,c.70G>A(E3)(p.24,V>I),确诊FPLD3。
PPARG是细胞核受体超级家族的一员,在脂肪前细胞向脂肪细胞分化的晚期大量存在,主要作用是识别各种激素和脂肪生成的相关受体配体,产生下游信号调控与脂肪细胞分化相关基因的表达,促进细胞分化的完成,维持成熟脂肪细胞的最终分化形态。PPARG基因突变通过干扰正常基因的表达或由于单倍体数量不足从而影响PPARG的表达[13],引起脂肪细胞生成及分化障碍,导致血中三酰甘油浓度过高,产生大量游离脂肪酸,超出白蛋白的结合能力,过多的游离脂肪酸会对胰腺腺泡细胞产生毒性作用,通过细胞膜脂质过氧化反应,损伤胰腺腺泡细胞,造成胰腺的缺血坏死,导致急性胰腺炎。
CGL患者的脂肪丢失可发生在生长发育的任何阶段,目前只能通过整容手术来改善外观,因此该病治疗的目的主要是纠正代谢紊乱及祛除导致脂肪异常分布的原因[14]。临床研究[15-16]表明,生长激素类似物可以增加机体肌肉的含量,减少内脏脂肪、降低三酰甘油、升高高密度脂蛋白,且不会影响胰高血糖素的水平。
综上所述,FPLD3型患儿临床表现主要为儿童、青少年或成年早期的对称部位脂肪缺失,以四肢和臀肌为主,同时可合并多种代谢紊乱,包括显著的胰岛素抵抗、高三酰甘油血症、胰腺炎的反复发作、多囊卵巢综合征和高血压,目前尚无特效治疗方法,对疑似患儿,详细询问家族史,尽早进行基因分析诊断,有助于发现未出现症状的FPLD3型患者。
利益冲突所有作者均声明不存在利益冲突
