CF3CH=CH2与大气Cl原子反应机理研究
2018-11-22刘艳
刘 艳
(1.渭南师范学院化学与材料学院,陕西渭南714099;2.陕西师范大学化学化工学院,西安710062)
氟利昂(CFCs)及氟氯代烷烃(HCFC)具有低沸点、热稳定性良好等物理和化学性质,曾被广泛应用在制冷剂和清洗剂生产等工业领域。然而,该类气体在大气中光解产生的Cl原子能破坏同温层中臭氧层,是导致高空臭氧层空洞的污染源之一,同时还会引发温室效应,因此CFCs及HCFC已被禁止使用。[1-4]众多的科学研究者致力于寻找可被环境接受的物质作为氟利昂与氟氯代烷烃的替代品。CF3CH=CH2(3,3,3-三氟丙烯,HFO-1243zf)是一种重要的氢氟烯烃,美国环境保护署(EPA)已经提出将CF3CH=CH2作为CF3CFH2(HFC-134a)在移动空调机组中的替代品[5]。但是,CF3CH=CH2分子中不饱和C=C双键,可通过加成、环氧化及羰基化等反应与大气中的活性自由基发生反应。因此,在大规模使用之前,必须考察其与大气自由基的反应机理及生成的后续产物是否对大气形成二次破坏。Papadimitriou[6]等研究了CF3CH=CH2与大气中Cl原子和NO3反应的速率常数。Nakayama[7]和Bernabe[8]分别从实验方面研究了CF3CH=CH2与Cl原子的反应速率及产物信息,提出如下初步反应机理:
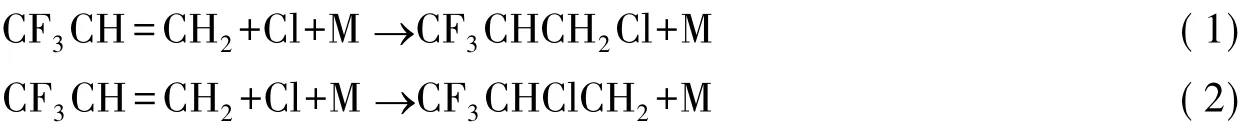
根据提出机理发现,CF3CH=CH2与Cl原子的初步反应为:Cl原子可分别加成到C=C双键两侧的C原子上,形成中间产物CF3CHCH2Cl和CF3CHClCH2。但是关于CF3CH=CH2与Cl原子的反应机理及微观动力学性质目前还未见文献报道,本文将从理论上采用密度泛函理论(DFT)研究气相中CF3CH=CH2与大气Cl原子的微观反应机理及可能产物,预测CF3CH=CH2与大气自由基间反应中可能存在产物及反应速率,这些研究成果对了解氟利昂替代品在大气中的反应性能具有理论指导意义。
1 计算方法
采用密度泛函理论[9-10]B3PW91/6-311++g(d,p)方法研究 CF3CH=CH2与 Cl原子的反应机理,给出了比较详细的势能面信息,对标题反应所有的反应物、中间体、产物和过渡态的几何构型进行了优化,通过分析振动频率确认了各驻点构型,并用内禀反应坐标[11-12](IRC)方法计算证实了各过渡态与相应反应物、中间体或产物的正确性。通过B3PW91水平零振动能(ZPE)校正后获得了反应势能剖面。计算工作均利用 Gaussian 09[13]程序完成。
采用传统过渡态(TST)计算最佳的反应通道在200 K至2 000 K温度下的速率常数,所用公式为:

其中:kB是 Boltzmann 常数(1.380 649 8×10-23J/K),h 是 Planck 常数(6.626 069 6×10-34J·s),c0是标准校正(2.687 19#1019molecules/cm3),R是摩尔气体常数,ΔG≠是决速步活化吉布斯自由能。
2 结果与讨论
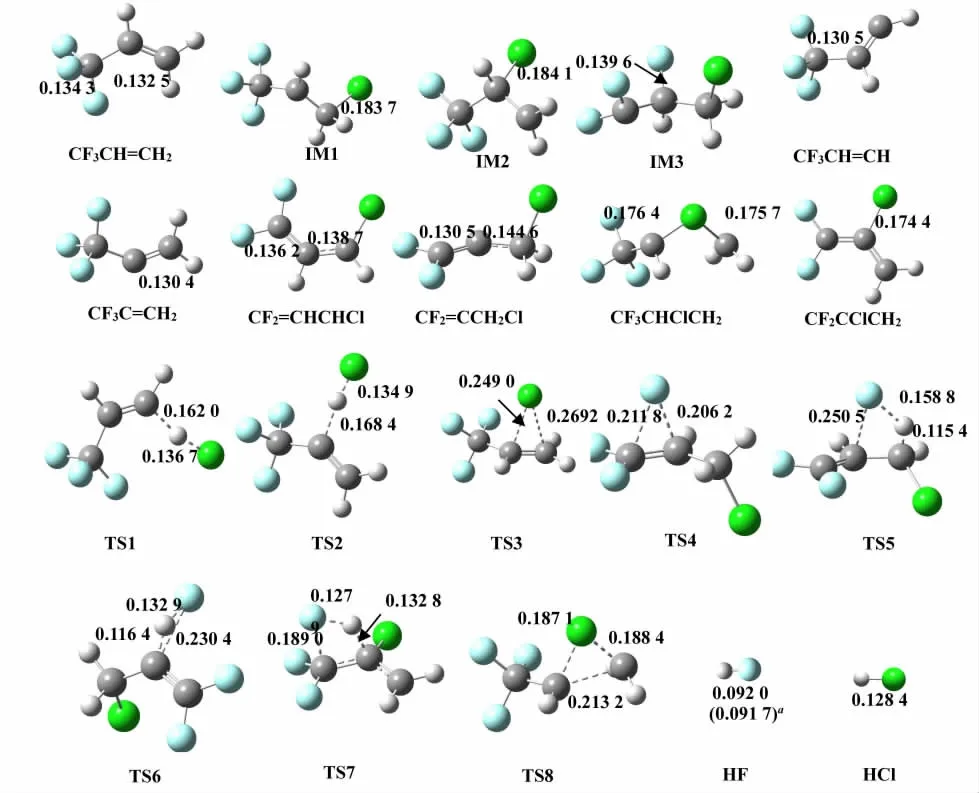
图1 B3PW91/6-311++g(d,p)水平得到的各物种几何构型(键长:nm,a实验数据[14])
B3PW91/6-311++g(d,p)水平优化得到的反应物、中间体(IM)、过渡态(TS)和产物的几何构型参数及部分实验值(见图1),其中:HF分子中H—F键长为0.092 0 nm,与实验值0.091 7 nm非常接近[14]。可见本文优化结果与实验值基本一致,表明所选用的计算方法是可靠的。所有反应物、中间体和产物的振动频率皆为实频,是反应势能面上的稳定点,所有过渡态均有且仅有唯一的虚频,表明它们分别是反应势能面上的一阶鞍点。通过IRC计算确认了各过渡态和对应反应物、中间体或产物的连接性,说明所有物种和过渡态均位于正确的反应路径上。
CF3CH=CH2与Cl原子发生反应得到6条可能的反应通道,得到6种产物,其中通道1与通道2为直接抽氢反应机理,而通道3至通道6为加成—消去机理。为了方便讨论,产物CF3CH=CH+HCl、CF3C=CH2+HCl、CF2=CHCHCl+HF、CF2=CCH2Cl+HF、CF2=CClCH2+HF 和 CF3CHClCH2分别记为通道 P1 至通道P6。图2为B3PW91/6-311++g(d,p)+ZPE在水平面上得到的势能剖面图。具体反应流程如下:
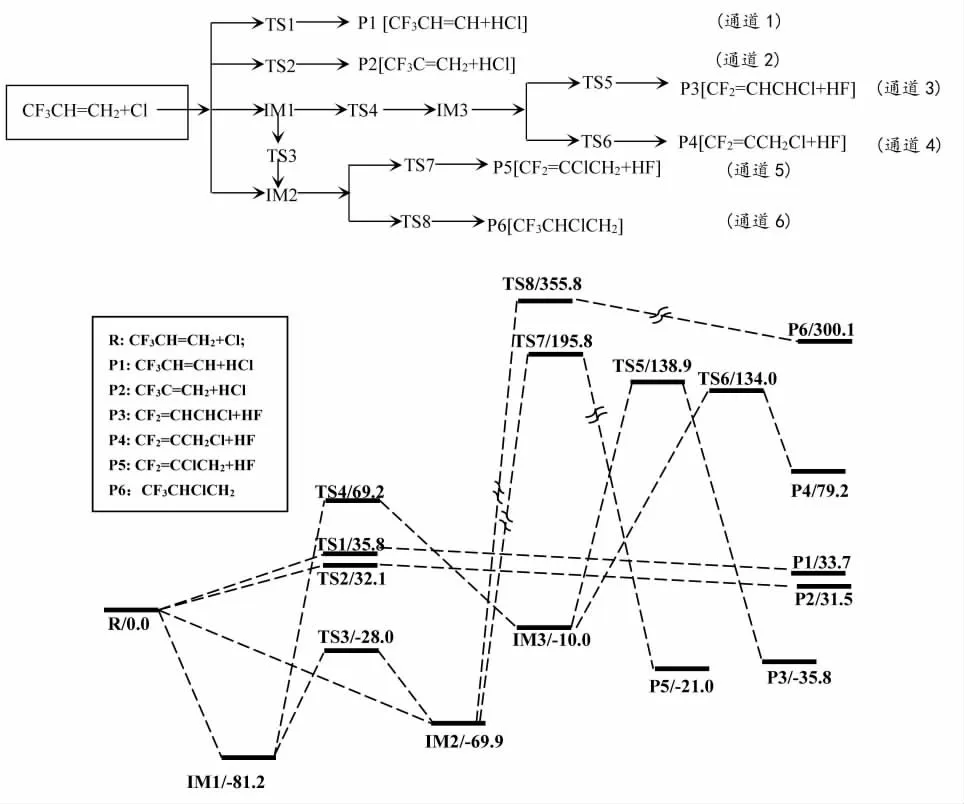
图2 反应在B3PW91/6-311++g(d,p)+ZPE水平上的势能剖面图(kJ/mol)
2.1 反应机理讨论
2.1.1 直接抽氢机理
Cl原子可与CF3CH=CH2分子中不同原子位上的H原子发生抽氢反应,分别经过渡态TS1和TS2,生成产物P1和P2。通道1中,Cl原子进攻CF3CH=CH2中C=C双键端基H原子,当Cl与H间的距离减小到0.136 7 nm时,形成过渡态TS1。在TS1中,欲断裂的C—H键长由反应物CF3CH=CH2中的0.108 4 nm拉长为0.162 0 nm,比反应物中C—H键长了0.053 6 nm。TS1的虚频为-240.43 cm-1,分析虚频振动模式可以看出,被进攻的H原子在C原子和Cl原子之间摆动。可见,该过渡态虚频的振动模式与相应过渡态走向反应物或产物的位移向量相对应。TS1的能垒高度相对于反应物为35.8 kJ/mol。接着,Cl原子携带H原子离去并生成裂解产物P1(CF3CH=CH+HCl),释放能量仅为2.1 kJ/mol。通道2中,Cl原子进攻反应物CF3CH=CH2中C=C双键内部H原子,当Cl与H间的距离减小到0.134 9 nm时,形成过渡态TS2。在TS2中,欲断裂的C—H键长拉长为0.168 4 nm,比反应物中C—H键长了0.060 0 nm。形成TS2需要越过的势垒高度为32.1 kJ/mol,比TS1的能垒仅低3.7 kJ/mol,产物P2的能量与P1也仅差3.7 kJ/mol。可见,通道1和2为两条竞争反应通道。
2.1.2 加成—消去机理
Cl原子可不经过过渡态,直接加成于CF3CH=CH2分子中双键两侧,分别生成中间体IM1和IM2,释放81.2和69.9 kJ/mol能量。可见,IM1比IM2更为稳定,Cl原子更易加成到端基C原子上。这一计算结果与Nakayama[7]和Bernabe[8]等人提出的初步反应机理相同。IM1和IM2中新生成的C—Cl键键长分别为0.183 7和0.184 1 nm。IM1和IM2能量随着C—Cl键长变化情况见图3。由图3可知,随着C—Cl键长不断增加,IM1和IM2能量亦不断增加,不存在能量最大点。因此,IM1和IM2的生成确实不经过任何过渡态,可直接生成。IM1可经过Cl原子迁移过渡态TS3,使得Cl原子由端基C上迁移至内侧C原子上,形成IM2。由IM1转化为IM2的能垒为53.2 kJ/mol,而逆反应的能垒高度为41.9 kJ/mol。

图3 IM1和IM2能量随C—Cl键长变化图
IM1也可经过F原子迁移过渡态TS4使得-CF3基团上的一个F原子迁移到内部C原子上,形成中间体IM3。TS3为一个三元环过渡态,其中欲断裂和生成的两个C—F键键长分别为0.269 2和0.249 0 nm。该反应具有较高能垒高度,为150.4 kJ/mol,且为吸热反应。生成的IM3的能量相对于IM1高出了71.2 kJ/mol。中间体IM3可经两条消去反应通道生成P3和P4(即通道3和4)。通道3和4中,IM3中迁移后的F原子可分别与端基和内侧H原子发生耦合消去HF反应,生成CF2=CHCHCl和CF2=CCH2Cl。所需经过渡态为TS5和TS6。其中,TS5为四元环过渡态,其中欲断裂的C—F和C—H键长分别为0.250 5和0.115 4 nm,而欲生成的F—H键长为0.158 8 nm。TS6为三元环过渡态,欲断裂的C—F和C—H键长分别为0.230 4和0.116 4 nm,而欲生成的F—H键长为0.132 9 nm。但是,TS5和TS6的能垒高度较高,分别为148.9和 144.0 kJ/mol。综合分析,通道 3和 4为多步反应通道,所需的两步能垒分别为 150.4、148.9 kJ/mol和150.4、144.0 kJ/mol,其速控步骤能垒均为150.4 kJ/mol。与一步反应通道1 和2的能垒高度35.8和32.1 kJ/mol相比,显然为不可行反应通道。
中间体IM2亦存在两条反应通道(即通道5和6),其中通道5中,IM2经过TS7发生HF消去机理。而通道6中,IM2中的Cl原子经过TS8,发生异构反应使得Cl原子插入C—C键之间,形成桥位Cl原子。但是这两步反应的能垒极高,其中TS8的能垒高达425.7 kJ/mol。因此通道5和6也是不可行的反应通道。
2.2 速率常数的计算
表1表示了用传统过渡态理论得到的反应通道1和2在200 K至2 000 K温度区间内的速率常数,分别用k1和k2表示(近似的认为吉布斯自由能是不随温度的变化而改变的)。由表1可知,k1和k2在同一温度时数值非常接近,这与其具有近似的势能一致。随着温度的升高,k1和k2不断增大。由此可见,高温能够加速反应的发生,而且随着温度的升高,k1和k2差值不断减小。例如,当温度在298 K时,k1在总速率中的分支比是0.11;当温度增加到2 000 K时,k1与k2接近的分支比不断接近,将增加到0.45,这说明在高温阶段,竞争更加激烈。

表1 200 K至2 000 K温度区间内的k1和k2(cm3·molecule-1·s-1)随温度的变化及其分支比
3 结语
用B3PW91/6-311++g(d,p)计算方法研究了CF3CH=CH2与Cl原子的反应机理,获得6条反应通道。结果表明:Cl原子可经过较低过渡态TS1和TS2抽提CF3CH=CH2分子中端基和内侧H原子,生成主要产物P1[CF3CH=CH+HCl]和P2[CF3C=CH2+HCl]。过渡态能所需的垒高度仅为35.8和32.1 kJ/mol,相差仅有3.7 kJ/mol,为竞争反应通道。两条反应通道的速率常数均随温度的升高而增大,呈正温度系数效应,且高温阶段,竞争更加激烈。
