浅谈物理化学中过渡态的搜索方法
2021-09-26张欣贺培楠
张欣,贺培楠
北京化工大学化学学院,化工资源有效利用国家重点实验室,北京 100029
1 引言
物理化学是化学类专业学生本科阶段的一门学位课程。化学动力学是研究化学反应中速率和机理的理论[1],是物理化学学习中的重要内容。化学动力学的发展先后经历了碰撞理论、过渡态理论和单分子反应理论等。其中,过渡态理论是目前研究最多、应用范围最广的化学动力学理论。对过渡态所处的“旧键未完全断裂,新键未完全形成”微观状态的理解是整个化学动力学学习的难点。因此,采用多种手段搜索过渡态,并直观、清晰地展示出温度对反应速率常数的影响,对物理化学相关知识点的学习具有重要意义[2,3]。
本文中,我们以1,3-丁二烯与乙烯的环加成反应为例,从微观角度出发,通过计算化学软件演示TS、QST2、QST3三种关键词搜索过渡态的过程,向学生直观地展示异构反应过渡态的立体构型;通过计算不同温度下的反应速率常数,揭示温度对反应速率的影响程度,帮助学生深入理解反应机理以及化学反应的本质。
2 实施过程
狄尔斯-阿尔德反应(Diels-Alder reaction)是有机合成中的一类重要反应,通常用于C―C键的形成。其中1,3-丁二烯和乙烯的反应是最典型的狄尔斯-阿尔德反应,其方程式如图1所示。

图1 1,3-丁二烯和乙烯反应制环己烯
本文以1,3-丁二烯与乙烯的环加成反应为例,通过Gaussian16[4]、GaussView6.0[5]等计算化学软件和Origin等数据处理软件,在B3LYP[6,7]/6-311+G(d,p)[8,9]水平下,实现多手段的反应过渡态搜索并获得宽温度范围的反应速率常数。具体过程可分为以下四步(图2):(1) 构建反应物和产物初始结构并进行几何优化,找到势能面上的稳定点,得到分子的能量;(2) 使用TS、QST2、QST3三种方法搜索过渡态,并对搜索到的过渡态进行频率计算和内禀反应坐标(IRC)验证;(3) 计算反应过程中热力学函数的变化,构建反应势能面,展示反应机理;(4) 根据(3)中得到的自由能垒,结合Arrhenius公式,揭示温度对反应速率常数的影响。

图2 计算化学方法辅助化学动力学学习流程图
3 过渡态的搜索与验证
3.1 反应物、产物分子的结构优化
构建反应物和产物结构并进行几何优化,寻找到能量局域最低的稳定点(能量对坐标的一阶导数为0)是搜索反应过渡态的第一步。我们通过GaussView6.0软件分别构建反应物(C4H6,C2H4)和产物(C6H10)的初始构型,并对其进行优化。如图3(a)、(b)所示,反应物分子1,3-丁二烯中的C=C键长(1.337 Å)大于乙烯的C=C键长(1.329 Å),产物分子环己烯的C=C键长(1.334 Å)相比1,3-丁二烯中的略短,但C―C键长有较大的差别。构型的改变对C=C以及C―C的键长影响较大。
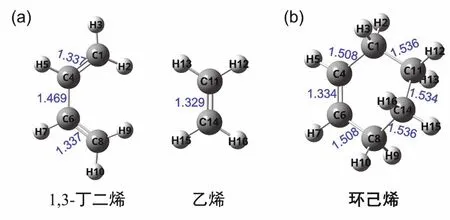
图3 反应物和产物分子的几何构型(键长单位为Å,1 Å = 0.1 nm)
3.2 三种方法搜索过渡态
Gaussian16提供了三种搜索过渡态的方法,分别通过三个关键词来执行:TS (输入文件中只需要提供过渡态的初猜结构)、QST2 (输入文件中需要提供反应物和产物结构)、QST3 (输入文件中需要提供反应物、产物结构和过渡态的初猜结构)。
3.2.1 关键词TS搜索过渡态
(1) 过渡态模型构建。为了能够顺利找到过渡态,首先需要给出一个合理的过渡态结构的初始猜测。如何对过渡态进行初猜?我们可以根据优化出的反应物和产物构型,结合过渡态的特点(旧键未完全断裂,新键未完全形成),对过渡态进行合理的初猜。在本例中,反应物分子发生环加成反应转变成产物主要是乙烯分子的C11和C14原子分别进攻1,3-丁二烯分子的C1和C8原子,同时C1=C4、C6=C8和C11=C14双键断裂,C4=C6双键形成(原子编号见图3(a))。因此我们可将过渡态初猜结构设定为C11和C14原子靠近C1和C8原子,并且拉长C1=C4、C6=C8和C11=C14双键,缩短C4―C6单键。
(2) 搜索过程。在使用GaussView6.0构建出过渡态的构型后,我们就可以使用Gaussian16软件中的TS关键词搜索过渡态。具体步骤如下:点击Calculate → Gaussian Calculation Setup,打开Gaussian的计算设置对话框,① 设置任务类型,选择Job Type → Opt + Freq (几何优化和频率计算),并选择优化到过渡态(TS(Berny)),选择Force Constants:Calculate at First Point,并且在下方的Additional Keywords里输入opt=noeigen,② 设置计算方法和基组等,选择Method → Method:Ground State、DFT、B3LYP,Basis Set:6-311+G(d,p),Charge:0,Spin:Singlet;③ 设置作业名称,选择Title →输入TS;④ 设置计算的chk文件名称,选择Link 0 → Options,设置Chkpoint File → Specify,TS.chk(计算生成的chk文件命名为TS.chk)。⑤ 设置完成后,点击Submit,保存输入文件,提交至Gaussian16程序计算。
3.2.2 关键词QST2搜索过渡态
(1) 输入文件的准备。将优化得到的反应物和产物结构写入同一个输入文件中,Gaussian程序会根据我们输入的反应物和产物结构自动生成一个过渡态的初始结构。打开GaussView6.0,粘贴优化好的反应物分子结构,点击File → Add to Molecule Group,粘贴优化好的产物分子结构。点击Tools → Atom List,打开原子列表编辑器,在Tag列修改原子序号,确保反应物和产物分子的原子序号一致。
(2) 搜索过程。点击Calculate → Gaussian Calculation Setup,打开计算设置对话框,设置任务类型Job Type → Opt + Freq,选择优化到过渡态(TS(QST2)),选择Force Constants:Calculate at First Point,并且在下方的Additional Keywords里输入opt=noeigen。方法和基组等其他设置同TS关键词搜索过渡态相同。设置完成后,点击Submit,提交至Gaussian16进行计算。
3.2.3 关键词QST3搜索过渡态
(1) 输入文件的准备。将优化得到的反应物、产物结构以及过渡态的初猜结构写入同一个输入文件中,Gaussian程序将根据输入的这三个构型重新生成一个过渡态的初始猜测结构。打开GaussView6.0,粘贴优化好的反应物分子结构,点击File → Add to Molecule Group,粘贴优化好的产物分子结构,继续点击File → Add to Molecule Group,构建过渡态的初猜结构。点击Tools → Atom List,打开原子列表编辑器,在Tag列修改原子序号,确保反应物、产物和过渡态分子的原子序号一致。
(2) 搜索过程。点击Calculate → Gaussian Calculation Setup,打开计算设置对话框,设置任务类型Job Type → Opt + Freq,选择优化到过渡态(TS(QST3)),选择Force Constants:Calculate at First Point,并且在下方的Additional Keywords里输入opt=noeigen。方法和基组等其他设置同TS关键词搜索过渡态相同。设置完成后,点击Submit,提交至Gaussian16进行计算。
图4(a)显示了TS方法优化后的反应过渡态的结构,C11原子进攻C1原子,同时C14原子进攻C8原子,C1、C4、C6、C8、C14与C11原子共同形成了六元环过渡态。将要断裂的C11=C14键长为1.389 Å、C1=C4和C6=C8键长为1.384 Å,将要形成的C4=C6键长为1.402 Å、C1―C11和C8―C14键长为2.246 Å。

图4 (a) 过渡态结构及虚频振动方向(键长单位为Å);(b) IRC曲线;(c) 环加成反应自由能面(括号内为电子能)
3.2.4 过渡态的确认
(1) 频率分析。在反应过程中,势能面上有一最小能量路径(minimum energy path,MEP)连接着反应物、过渡态和产物的结构。过渡态是最小能量路径上的能量最高点,也是一阶鞍点,它的能量对坐标的一阶导数为零,在反应坐标方向上对坐标的二阶导数为负,在其他方向上为正。当振动频率仅有一个虚频时,就证明这是一个过渡态。使用GaussView6.0软件查看过渡态的频率计算结果以及频率的振动方向可知,振动频率中只有一个虚频(544.31icm−1),且虚频的振动方向对应于C1―C11和C8―C14的生成(图4(a))。以上分析结果进一步证明了搜索到的结构确实为环加成反应过渡态。
(2) 反应路径验证分析。为了验证该过渡态结构是连接反应物和产物的过渡态,我们进行了内禀反应坐标(IRC)计算。IRC计算从过渡态开始,根据能量降低的方向寻找过渡态连接的两个极小值,IRC是连接反应物、过渡态和产物的能量最低的反应路径。计算结果表明,IRC曲线上每个点都对应一个亚稳态结构(如图4(b)所示),能量最高点对应过渡态结构,曲线两端分别接近反应物和产物结构,从图中可以明显看出环加成反应过程伴随着C1―C11和C8―C14、C4=C6的生成以及C1=C4、C6=C8、C11=C14的断裂。
3.3 反应势能面的构建
反应物、过渡态和产物的电子能、内能、焓和吉布斯自由能值如表1所示。在298.15 K、101.325 kPa下,该环加成反应(C4H6+ C2H4→ C6H10)的ΔrHm= −145.34 kJ·mol−1,ΔrUm= −142.86 kJ·mol−1,ΔrGm=−82.11 kJ·mol−1,为热力学自发过程。图4(c)显示了B3LYP/6-311+G(d,p)水平下反应的吉布斯自由能面,反应物分子1,3-丁二烯和乙烯反应生成产物环己烯过程放热82.11 kJ·mol−1,反应物需要越过152.84 kJ·mol−1的能垒才能转变成产物。结合《物理化学》教材中的化学动力学原理可知,该反应在常温下的速率非常缓慢,需要升高温度才能有效提高环己烯的产率。

表1 B3LYP/6-311+G(d,p)水平下反应物、过渡态和产物的电子能E、内能U、焓H和吉布斯自由能G
3.4 温度对反应速率常数的影响
结合物理化学中的Arrhenius公式,热力学形式的反应速率常数的计算公式如下:

其中σ是反应路径的简并度,kB是玻尔兹曼常数,T是反应温度,h是普朗克常数,R是摩尔气体常数,P0是标准压力;Δn = n− 1,n是所有反应物的计量系数之和;ΔG≠是反应的活化吉布斯自由能,即过渡态和反应物的自由能之差。
通过3.3中的计算结果可知,298.15 K时,反应物到产物的活化吉布斯自由能ΔG≠为152.84 kJ·mol−1。因此,298.15 K时反应的速率常数为5.14 × 10−16(mol·m−3)−1·s−1。
此外,化学动力学理论表明升高温度有利于提升反应速率。那么,温度对该异构反应速率常数的影响有多大呢?通过计算不同反应温度下(373.15、423.15、473.15、523.15和573.15 K)的环加成反应速率常数(表2),我们发现随着反应温度的升高,自由能垒逐渐升高,且反应速率常数有5个数量级的提升(373.15 K–573.15 K)。

表2 不同温度下环加成反应的活化吉布斯自由能ΔG≠和反应速率常数k
4 结语
本文以1,3-二丁烯和乙烯的环加成反应中过渡态的搜索为例,介绍了三种搜索反应过渡态的方法及其详细操作步骤。本文不仅通过计算化学可视化软件将过渡态这一抽象的概念进行了直观展示,而且讨论了了温度对反应速率的影响,以期实现帮助学生深入理解化学动力学理论知识、激发对物理化学的学习兴趣、培养创新思维能力和科研能力的目的。
