重质有机资源热解过程中自由基诱导反应的密度泛函理论研究
2021-06-02毕山松郭啸晋王波徐祥赵丽凤刘清雅
毕山松,郭啸晋,王波,徐祥,赵丽凤,刘清雅
(1.中国科学院大学,北京100049;2.中国科学院先进能源动力重点实验室(工程热物理研究所),北京100190;3.中国科学院能源动力研究中心,江苏连云港222069;4.北京化工大学化学工程学院,北京100029)
热解反应几乎在所有重质有机资源的热转化过程中均会发生[1,2],因此,针对热解反应进行研究,不仅可帮助深入了解重质有机资源的热解机理、从而优化热解技术本身,而且可以对于其他热转化技术的优化提供支持。然而,从化学结构看,重质有机资源是由数以百计的官能团构成数万甚至数十万分子量的高分子聚合物,在热解过程中通常会发生超过20000个反应[3]。由于原料化学结构难以确定、反应体系庞大,因此,无法使用已成功应用于较简单体系的常规量子化学方法进行模拟。一个合适的策略是对反应物和反应种类进行集总[4,5],建立相对简单的反应体系,以减少计算量而实现预测。研究者通过对重质有机资源中不同共价键分布及其反应行为之间的关联研究[5−14],证实了以共价键作为着眼组分进行热解研究具备可行性。
在100余年的热解反应研究历史中,研究者通过种种实验证实了重质有机资源的热解反应以自由基反应为 主[2,15−19],即在热环 境 下,复 杂烃类的共价键首先发生断裂形成自由基,随后自由基作为活性组分发生后续反应而生成中间产物乃至最终形成产物。热解中的自由基反应遵从链式反应机制[1,17],即包含链引发、链传递和链终止三个过程。在上述三个过程中,链引发的反应物为共价键,而链终止反应的本质则是自由基相互反应形成新的共价键,链传递反应相比链引发和链终止反应更为复杂,其反应物为自由基和共价键,产物为新自由基和新共价键。一般认为,发生链引发过程共价键的布居数由该共价键的解离能和热解反应温度决定,而链终止过程则为自发过程,其能垒为0。
因此,基于共价键在热解过程中的“断裂-生成”[20−22](即链引发和链终止过程)机理,研究者[23,24]使用共价键为着眼组分建立Boltzmann-Monte Carlo-Percolation (BMCP)统计机理模型,对生物质和煤热解挥发物以及不同煤阶煤的热解行为进行预测,结果表明,在较高温度下,模型模拟的结果与实验结果较为接近,但是在低温下对热解产物产率预测误差偏高,这可能是由于该模型未考虑链传递反应使得低温下发生的反应速率被低估导致的。由于热解产生的大量挥发分中含有大量自由基,它们在非约束体系中会与共价键发生碰撞,发生诱导反应[2,17],使较强共价键在较低温度下即发生断裂,从而使重质有机资源在较低温度下即可发生反应。
本研究针对重质有机资源中的不同共价键与热解过程产生的典型小分子自由基之间诱导反应进行计算,确定未成对电子对共价键解离能的影响。其结果不仅可为BMCP模型精度的进一步提高提供支持,同时也为进一步探索温和热解条件下自由基链传递过程提供基础数据,从而为探索低温下热解反应的可能途径与建立反应集总模型提供基础数据。
1 计算模型与计算方法
1.1 计算模型
尽管重质有机资源组成千变万化,但若以共价键为着眼点,除去丰度不高、作用不大的N和S元素的相关共价键,在重质有机资源热解过程中发 生 反 应 的 共 价 键 有Car=Car、Car−Car、Cal−Cal、Car−Cal、Car−O、Car−H、Cal−O、Cal−H和O−H键 等九种。

表1 与自由基发生诱导反应的共价键Table 1 The covalent bonds induced by free radicals
需要说明,尽管轻质有机资源和重质有机资源的区分为分子量,但一般认为,重质有机资源和轻质有机资源最主要的区别在于其芳香结构的多寡。例如对石油而言,脂肪烃组成的汽柴油等油品一般认为是轻质有机资源,而常压渣油和减压渣油等富含沥青质和胶质(其化学本质为缩合芳环结构)的油品则被认为是重质有机资源。芳香结 构 主 要 包 含Car=Car、Car−Car、Car−Cal、Car−O和Car−H键,其中,Car=Car和Car−Car键居于重质有机资源的芳香结构核心,相比居于侧链或芳香核心外围的其他七种共价键而言,这两种共价键的诱导反应位阻极大,实际较难发生,故不再考虑。因此,选取了50种有机物作为上述七种共价键的模型物,其发生诱导反应的共价键如表1所示。
1.2 计算方法
本研究选择6-31G**基组,使用四个物理核心运行Gaussian 09[25]程序,分别采用B2PLYP[26,27]、mPW2PLYP[28,29]、ωB97XD[30,31]、B3LYP-GD3[32,33]和M06-2X-GD3[34,35]等五种基于密度泛函理论的方法对八种有机物(见附表1)中Cal−Cal、Cal−O、Cal−H和O−H键的解离能进行计算。综合考虑计算精度和工作效率以判定不同方法的优劣。计算精度CA通 过式(1)计算得到:

式中,BDE为 通过计算得到的键解离能;BDEref为同种共价键参考解离能,来源于键能数据手册[36]。
计算耗时与计算精度结果如图1所示。由图1可知,两种双杂泛函B2PLYP和mPW2PLYP的精度较其他三种普通泛函为高,精度分布较为密集,但计算耗时较长;在三种普通泛函中,ωB97XD与前面两种双杂泛函的精度更为接近,精度分布较B3LYP-GD3、M06-2X-GD3更为密集,且计算耗时与两者相当。因此,选用ωB97XD/6-31G**计算级别进行后续的研究。

图1 五种方法的计算耗时与计算精度Figure 1 The calculation time and accuracy of five methods
参考相关文献[37],构建自由基诱导共价键断裂反应的反应物、过渡态及产物的分子模型。随后通过优化和频率计算获得它们的几何结构和分子振动频率,并确认反应物与产物无虚频、过渡态有且只有一个虚频。最后对过渡态进行内禀反应坐标计算(IRC)以保证过渡态与反应物和产物的正确衔接。
2 结果与讨论
2.1 甲基自由基对不同共价键的诱导作用
为了研究·CH3对不同共价键的诱导作用,对·CH3在典型热解温度500℃下诱导七种共价键反应过程的Gibbs自由能(Gibbs energy,G)进行了理论计算。反应过程量变化示意图如图2所示。·CH3诱导七种共价键的自由能能量变化如图3所示。

图2 自由基诱导共价键的能量变化示意图Figure 2 Schematic diagram of the free energy change of induction reaction between covalent bond and radical
同时,为了说明诱导反应发生的难易程度与复杂程度,定义能垒变化范围(the range of energy barrier,REB)为相同自由基诱导同种共价键反应能垒的最小值到最大值。同样,定义了反应自由能 变 化 范 围(the range of delta G,RDG)。·CH3诱导七种共价键诱导反应的REB和RDG列于表2。
同种自由基与七种共价键发生诱导反应,能垒的高低可以反映共价键因诱导断裂的难易程度,即自由基诱导不同共价键断裂的能力强弱。根据表2的数据,可以得到·CH3诱导不同共价键断裂的能力强弱顺序为:O−H>Cal−H≈Car−H>Cal−O>Cal−Cal>Cal−Car≈Car−O键。由表2数据可知,考虑重质有机资源中的常见桥键Cal−Cal和Car−O键,其反应能垒由原有解离能约300 kJ/mol可降至<200 kJ/mol,相同布居数对应的反应温度可降低150 K以上。
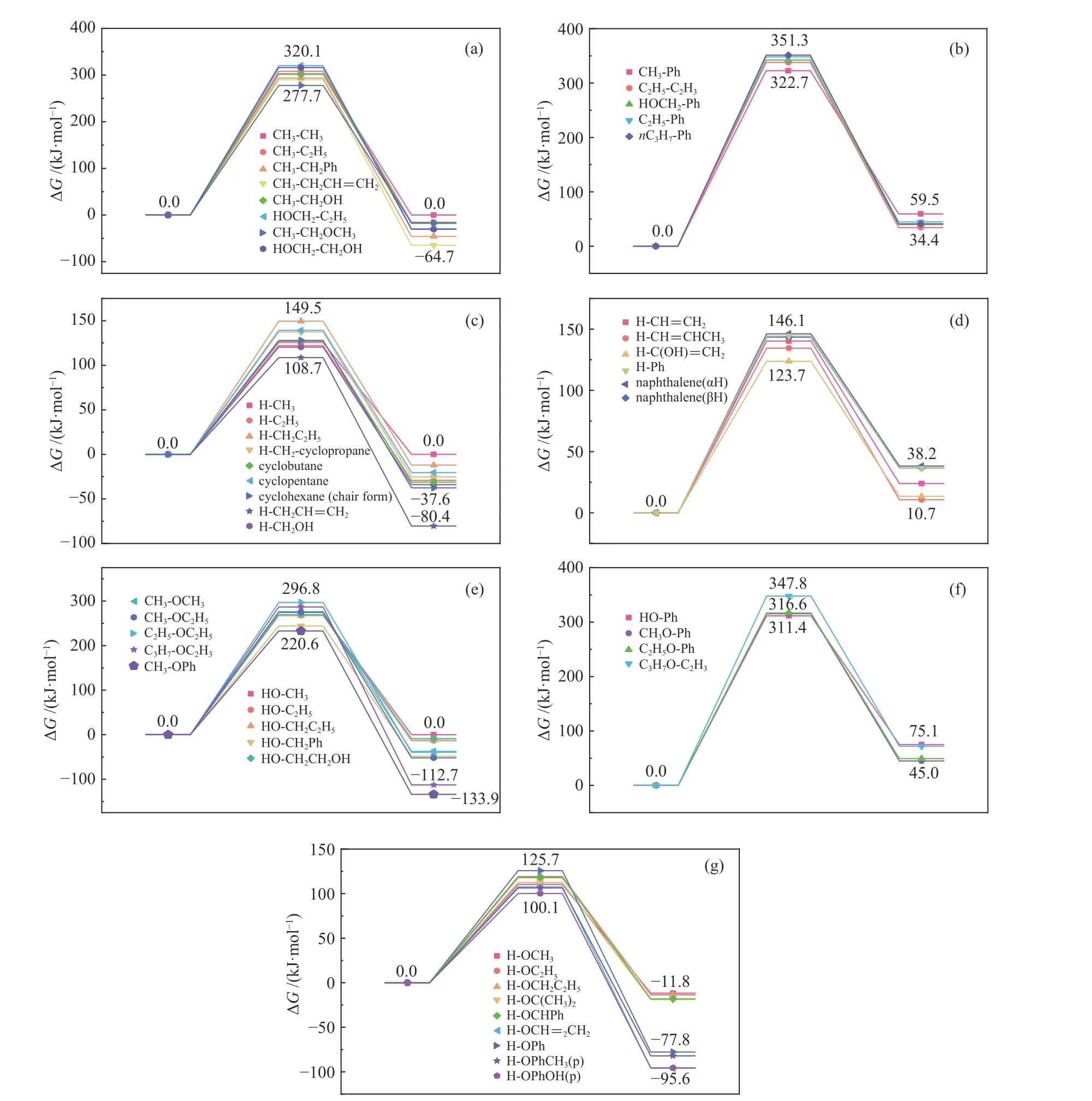
图3 ·CH3诱导七种共价键的能量变化Figure 3 Free energy change of seven covalent bonds induced by ·CH3
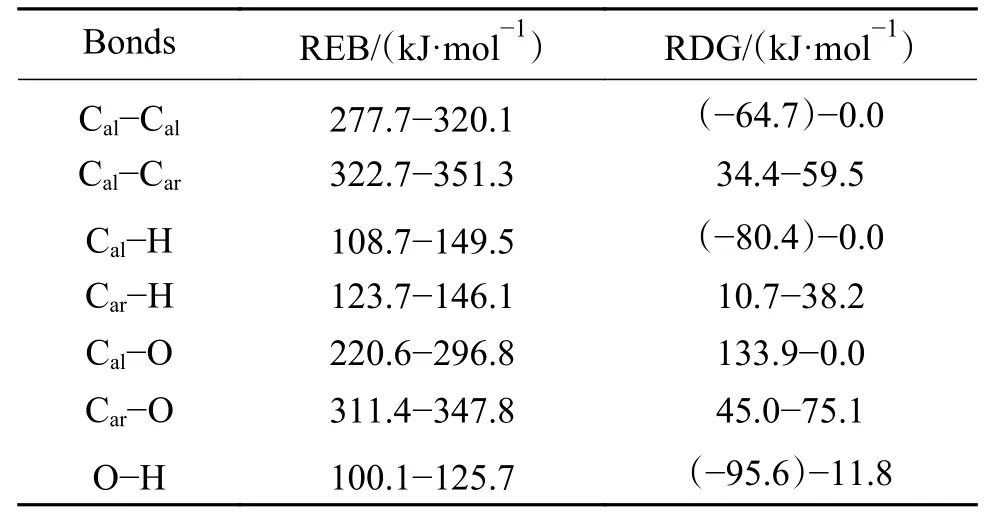
表2 ·CH3诱导七种共价键的能垒范围和Gibbs能变化Table 2 Range of energy barrier and the change of delta G of seven covalent bonds induced by ·CH3
由于微观尺度热解过程中的基元反应可逆,因此,在恒温恒压条件下,ΔG<0方向的反应在热力学上具有优势而更容易发生,以图2为例,在实际热解过程中更容易发生产物向反应物的逆反应。根据表2的数据可以发现,·CH3诱导Car−H、Cal−Car和Car−O键的反应ΔG>0,因此,在实际热解过程中,·CH3主要诱导O−H、Cal−H、Cal−O和Cal−Cal键断裂。
由表2可知,·CH3诱导Cal−H和Car−H键的能垒分别在110−150、120−145 kJ/mol,差别不大,这说明C−H键中C原子种类对·CH3诱导C−H键断裂的能垒影响并不明显;·CH3诱导Cal−Car和Car−O键的能垒分别在320−350、310−350 kJ/mol,相差不大,这说明·CH3诱导Car-X(X=O,Cal)断裂形成含Car烃基的能垒受X基团的种类的影响不大。
此外,还可发现,·CH3诱导Car−H键的能垒比诱导Car−Cal键的能垒约低三倍,这说明反应空间位阻效应[16,38]对能垒有较大影响,Car所连接的基团体积越小,诱导能垒越低、反应越容易发生;Car甚至Cal所连接的基团体积相差不大时,反应能垒与基团的种类基本无关。
2.2 羟基自由基对不同共价键的诱导作用
为研究·OH对不同共价键的诱导作用,对·OH在500℃下诱导七种共价键反应过程的Gibbs自由能进行了理论计算。·OH诱导七种共价键的自由能能量变化如图4所示,七种共价键诱导反应的REB和RDG列于表3。

图4 ·OH诱导七种共价键的能量变化Figure 4 The free energy change of seven covalent bonds induced by ·OH
根据表3的数据可以得到,·OH诱导不同共价键断裂的能力强弱顺序为:O−H>Cal−H≈Car−H>醚 中Cal−O≈Cal−Cal>Cal−Car≈醇 中Cal−O>Car−O键。考虑到ΔG<0方向的反应更容易发生,因此,在实际热解过程中,·OH主要诱导O−H、Cal−H、Car−H、醚中Cal−O和Cal−Cal键断裂。

表3 ·OH诱导七种共价键的能垒范围和Gibbs能变化Table 3 The range of energy barrier and the change of delta G of seven covalent bonds induced by ·OH
根据表3可以发现,·OH诱导Cal−O键时,REB和RDG分成醚和醇两种情况。但·OH诱导醇中Cal−O键断裂具体路径是其通过诱导HO−Cal断裂,而诱导醚中Cal−O键断裂具体路径是其通过诱导RO−Cal断裂,这说明位阻效应不显著时,·OH诱导共价键中与其相同基团(−OH),即发生·OH的同基团诱导交换反应,存在着较强的排斥作用而使诱导能垒升高。
同样,对于在基团位阻效应不显著的情况下,·OH诱导酚中Car−O键断裂的能垒比醚中Car−O断裂的能垒高。因此,同基团效应也是影响·OH诱导不同共价键断裂的能力强弱的因素,解释了前面的·OH诱导能力强弱顺序。
2.3 氢自由基对不同共价键的诱导作用
为研究·H对不同共价键的诱导作用和反应发生的方向性,对·H在500℃下诱导上述七种共价键过程的能量进行了理论计算。·H诱导七种共价键的自由能能量变化如图5所示,七种共价键诱导反应的REB和RDG列于表4。
根据表4的数据可以得到,·H诱导不同共价键断裂的能力强弱顺序为:O−H≈Cal−H>Car−H>Cal−O>Car−O≈Cal−Cal>Cal−Car键。考虑 到ΔG<0方向反应更易发生,因此,在实际热解过程中,·H主要诱导除了Car−H之外的O−H、Cal−H、Cal−O、Car−O、Cal−Cal和Cal−Car键断裂。
此外,平均上来看,·H对不同共价键的REB最高,即·H对共价键解离能的降低作用最为显著,考虑到自由基诱导反应主要作用为未成对电子,与该电子所处原子种类关系不大,因此,可以推断,诱导反应能垒的不同主要是空间位阻不同所致,相比·CH3和·OH,·H具有最小的体积,能以较低能量逼近势能面极值,使反应能垒较低而使得REB较高。
2.4 自由基诱导反应对共价键解离能的影响
由上述分析可知,尽管自由基对共价键的诱导作用会显著降低反应能垒,使反应在较低温度下即可发生,但单一自由基对共价键解离能的影响似无共性规律。为进一步分析自由基诱导对共价 键 解 离 能 的 影 响,将Cal−Cal、Cal−Car、Cal−H、Car−H、Cal−O、Car−O和O−H等共价键模型物的键解离能和与不同自由基发生诱导反应的能垒数据进行汇总并列于表5。需要说明,自由基有效作用的距离不尽相同,但差异不大,造成诱导反应能垒差异的主要变量应为不同自由基和共价键之间的空间位阻效应。自由基作用的有效距离见附表2−4。
对比表5中BDE和能垒数据可发现,同种自由基诱导反应的能垒变化不大,例如Cal−Cal的BDE约 为360 kJ/mol,·CH3、·OH、·H诱 导Cal−Cal键的能垒分别约为310、250、230 kJ/mol;又如·OH诱导Cal−H、Car−H键的能垒分别为77、87 kJ/mol,诱导Cal−O−H、Car−O−H键的能垒分别为71、67 kJ/mol。尽管共价键解离能值差异较大,但自由基诱导反应能垒则相对较为集中[39],且当共价键空间位阻作用类似而解离能不同时,自由基种类引起的差异大于共价键种类不同引起的差异。这说明空间位阻效应在自由基诱导反应中起主导作用。
进一步分析可知:
第一,对比·CH3对C−H键和·OH对C−H键的诱导、对比·OH和·H对C−C键的诱导、以及对比·CH3对C−O键和·H对C−O键的诱导,即当排除·OH和·H的同基团诱导交换反应时,不同自由基诱导共价键反应能垒高低顺序为EB(·CH3)>EB(·OH)>EB(·H),这 与 诱 导 自 由 基 体 积 大 小 有关,总体而言,·OH诱导能垒比·H的高约40 kJ/mol,·CH3比·OH、·H的 诱 导能 垒 差 分 别 高 约 为50、90 kJ/mol,三种不同自由基的诱导能垒差值具有可加性,这说明计算结果可以自洽,也充分说明微观反应计算出的能垒为状态函数,这也从结果角度证明了计算方法和计算精度可信。
第二,当存在·OH或·H的同基团诱导交换反应时,对应的反应能垒升高,例如·OH诱导醇中Cal−O断裂的反应能垒高于其他两种自由基诱导能垒,再如·H诱导三种含H共价键(Cal−H、Car−H和O−H)断裂的反应能垒高于其他两种自由基诱导能垒;定量来看,以·OH诱导C−O键断裂为例,当假设不存在同基团诱导交换反应的能垒提高时,按照第一中的原则可算出·OH诱导能垒应约为220(Cal−O键)和260 kJ/mol(Car−O键),而实际诱导反应能垒则分别约为220和330 kJ/mol,由此可得同基团诱导交换反应导致的能垒提高值约为70 kJ/mol,其他计算结果也与此值一致。该结果反映了同基团间的相互作用导致不同自由基诱导共价键反应能垒差不再恒定,因此,在计算时还需考虑自由基中心原子是否与共价键相连的原子一致,并因此修正相应诱导反应能垒。

图5 ·H诱导七种共价键的能量变化Figure 5 The free energy change of seven covalent bonds induced by ·H
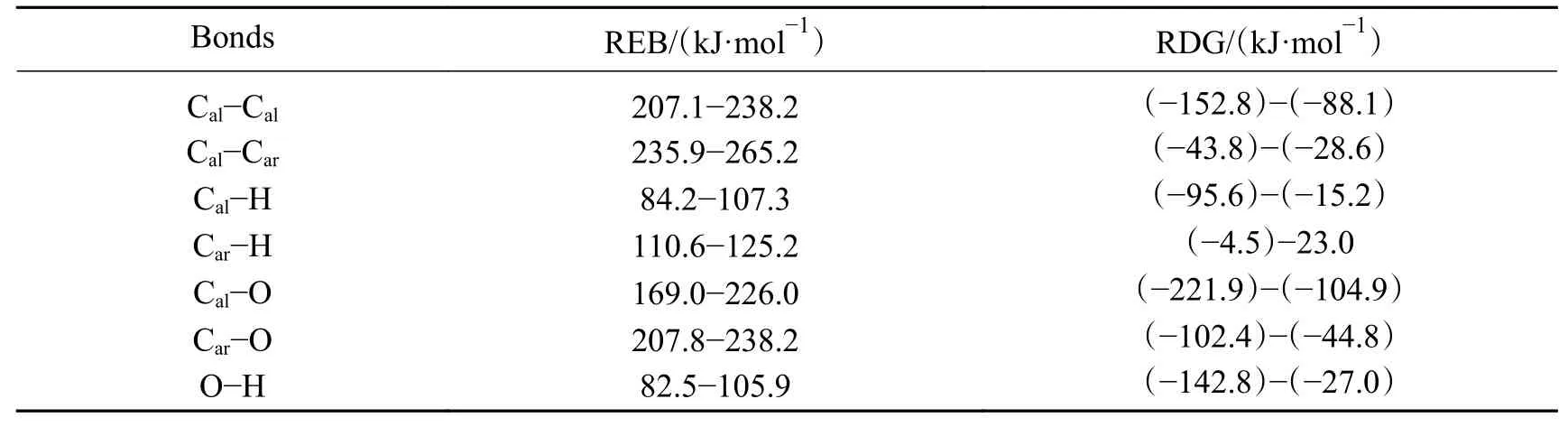
表4 ·H诱导七种共价键的能垒范围和Gibbs能变化Table 4 The range of energy barrier and the change of delta G of seven covalent bonds induced by ·H

表5 共价键的键解离能及诱导反应能垒Table 5 The BDEs and the energy barriers of the induction reaction
3 结论
空间位阻效应对自由基诱导反应的能垒的影响占主要地位,共价键种类的影响相对次要。
不存在·OH和·H的同基团诱导交换反应时,·OH诱导能垒比·H的高约40 kJ/mol,·CH3比·OH、·H的诱导能垒分别高约为50、90 kJ/mol;
存在·OH或·H的同基团诱导交换反应时,会导致能垒约有70 kJ/mol的提高,在计算时应判断诱导反应的具体情况并加以修正。
在缺少不同诱导反应的能垒数据时,可以利用上述值进行估算,此结论可在一定范围内推广。
