AQP4抗体及MOG抗体双阳性合并甲状腺功能异常1例NMOSD报道及文献复习
2018-10-12蒋继浩
蒋继浩
视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disorders,NMSOD)是一组临床少见的中枢神经系统自身系统免疫性脱髓鞘性疾病。临床上根据血液中AQP4抗体检出情况被分为AQP4抗体(+)和AQP4抗体(-)两组。研究发现,在一些AQP4抗体阴性的患者体内可检测到髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoproeein,MOG)抗体。临床上少有报道AQP4抗体及MOG抗体均阳性的病例。本文介绍1例复发缓解型NMOSD患者,血液检查时发现AQP4抗体及MOG抗体均阳性并合并亚临床甲亢等自身免疫病。
1 病例摘要
患者,女,33岁。6 y前曾患顽固性呃逆、呕吐3、4 m后好转;2 y前双下肢麻木无力,治疗好转;1.5 y前左眼视物不清治疗好转;再发1 w后于2018年1月9日入院。患者于6 y前(2012年1月)产后50 d左右出现顽固性呃逆、呕吐,进食后明显,至当地医院消化科就诊,消化系统检查未见明显异常。后考虑“产后抑郁”在当地精神病院治疗(具体不详),好转不明显,症状持续3~4 m后逐渐消失。2 y多前(2015年3月)有“麻疹疫苗接种史”,约1 m后突发头部及颈部疼痛,伴有大小便费力,后逐渐出现胸部(约双乳头水平)束带感及以下麻木、双下肢无力,行走费力,需家人搀扶。当时无视物不清,无双上肢麻木无力,无呼吸异常。当地医院就诊,完善颈椎MRI示:“C2-C6水平异常信号”,诊断“脊髓炎”,给予激素冲击治疗(甲强龙1000 mg×5 d,500 mg×3 d,240 mg×3 d,120 mg×2 d,60 mg×7 d;后改为40 mg强的松口服,每周减5 mg至停药),并口服免疫抑制药1 m(硫唑嘌呤),用药期间大小便很快恢复正常,减药过程中肢体麻木无力逐渐消失。1 y多前(2016年7月)出现左眼视物不清及活动时眼球、眼眶疼痛,最严重时仅有光感。无肢体无力,无大小便异常,给予激素冲击治疗(用法同前)及球后激素注射1次(5 mg地塞米松),自2016年8月开始每月静点2次“环磷酰胺”每次0.4 g至现在,减药过程中视力逐渐恢复至正常。1 w前劳累后(2018年1月3日)再次出现左眼视物不清及活动时眼球疼痛,视物不清逐渐加重,4 d前左眼失明,此次发病2 d当地医院给予激素冲击治疗(甲强龙1000 mg×3 d,500 mg×3 d),入院时仅有光感,此次发病无肢体无力,无二便障碍。既往否认高血压病、糖尿病及心脏病等病史,否认肝炎、结核等病史,否认抽烟、饮酒史。父母健在,1姐患有甲状腺功能亢进。入院查体:体温36 ℃,脉搏88次/min,呼吸20次/min,血压125/80 mmHg。神志清,精神一般,言语流利,高级皮质功能未见明显异常,双眼运动灵活,无眼震,左眼直接对光反射迟钝,间接对光反射灵敏,右眼直接及间接对光反射灵敏,左眼仅有光感,双额纹对称,双侧鼻唇沟等深,伸舌居中,四肢肌力、肌张力正常,双侧腱反射对称,双侧Babinski征阳性,感觉、共济查体未见明显异常,脑膜刺激征(-)。入院前辅助检查:颈部MRI(2015年4月):C2-C6水平异常信号(见图1)。颈部MRI(2018年1月7日):较2015年颈髓内异常信号大部消失。头部MRI(2018年1月7日):左侧放射冠区点状脱髓鞘(见图2、图3)。入院后完善相关检查示:实验室检查:血常规:16.04×109/L,中性粒细胞计数12.63×109/L,中性粒细胞百分率78.7%,血小板、红细胞数、血红蛋白大致正常。甲状腺功能全项:促甲状腺激素0.13ulU/ml(0.34-5.6),游离三碘甲状腺原氨酸1.94 pg/ml(2.3-4.2),甲状腺摄取率53%(32.0-48.4),三碘甲状原氨酸、甲状腺素、甲状腺球蛋白抗体、甲状腺微粒体抗体大致正常。补体C3 0.50 g/l(0.79-1.52),补体C4 0.09g/L。抗链O抗体138 IU/ml(25-116)。尿便常规、肝肾功、电解质、血糖、血脂、凝血四项、D-二聚体、同型半胱氨酸、免疫球蛋白、C 反应蛋白、类风湿因子、乙肝表面抗原、丙肝抗体、梅毒艾滋抗体、抗核抗体谱、抗中性粒细胞抗体、抗心磷脂抗体未见明显异常。腰穿脑脊液检查:无色清亮透明,压力90 mmH2O,压颈实验通畅,CSF:细胞总数220×106/L,白细胞计数20*106/L,脑脊液蛋白、糖、氯、免疫球蛋白A/免疫球蛋白均正常。送检北京协和医院检查结果回报:血AQP-4-Ab阳性(+)1∶100,脑脊液AQP-4-Ab阴性;血 NMO-IgG (-) 、脑脊液NMO-IgG (-);脑脊液细胞学正常。送北京大学附属第一医院检查回报:BBB通透性=5.02×10-3(正常<5.0×10-3),血抗少突胶质细胞糖蛋白抗体(MOGAb)=0.728(正常<0.640),脑脊液抗少突胶质细胞糖蛋白抗体(MOGAb)=0.375(正常<0.560)。脑脊液寡克隆区带(OCB)阴性、IgG指数=0.73(正常<0.85)、脑脊液的IgG鞘内合成率(IgG-Syn)=2.62 mg/24 h(正常<7.0 mg/24 h)、脑脊液髓鞘碱性蛋白(MBP)=0.44 μg/L(正常<3.5 μg/L)、血髓鞘碱性蛋白(MBP)=2.02 μg/L(正常<2.5 μg/L)、脑脊液髓鞘碱性蛋白自身抗体(MBP. Ab)=0.190(正常<0.650)、血髓鞘碱性蛋白自身抗体(MBP. Ab)=0.544(正常<0.750)。视觉诱发电位(2018年1月15日):左侧P100未引出肯定波形,右侧P100潜伏期延长,波形分化不良。听觉诱发电位:听域:左:20Db,右:20Db,各波潜伏期及波形均未见异常。体感诱发电位:刺激右正中神经,各波均未见异常,刺激右胫神经,皮质未引出肯定波形,余各波均未见异常,提示中枢性异常。胸髓MRI平扫(2018年1月17日):胸6-7水平髓内异常信号(见图4)。患者入院后给予口服强的松及给予七叶皂甙、复方脑肽节苷脂等药物应用,并用免疫抑制剂吗替麦考酚酯,患者左眼视力逐渐恢复,于2018年1月19日出院,出院时左眼可看清1米左右指数。电话随访2018年1月29日左眼视力恢复至0.1,2018年3月17日左眼视力恢复至0.3,不影响日常生活及工作。
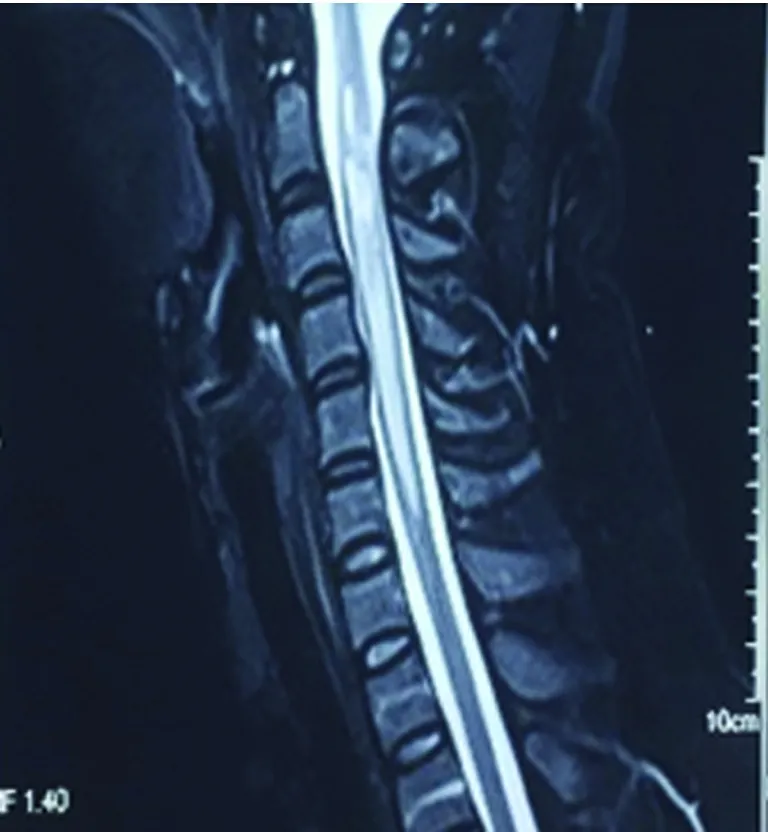
图1 颈部MRI可见C2-C6水平异常信号
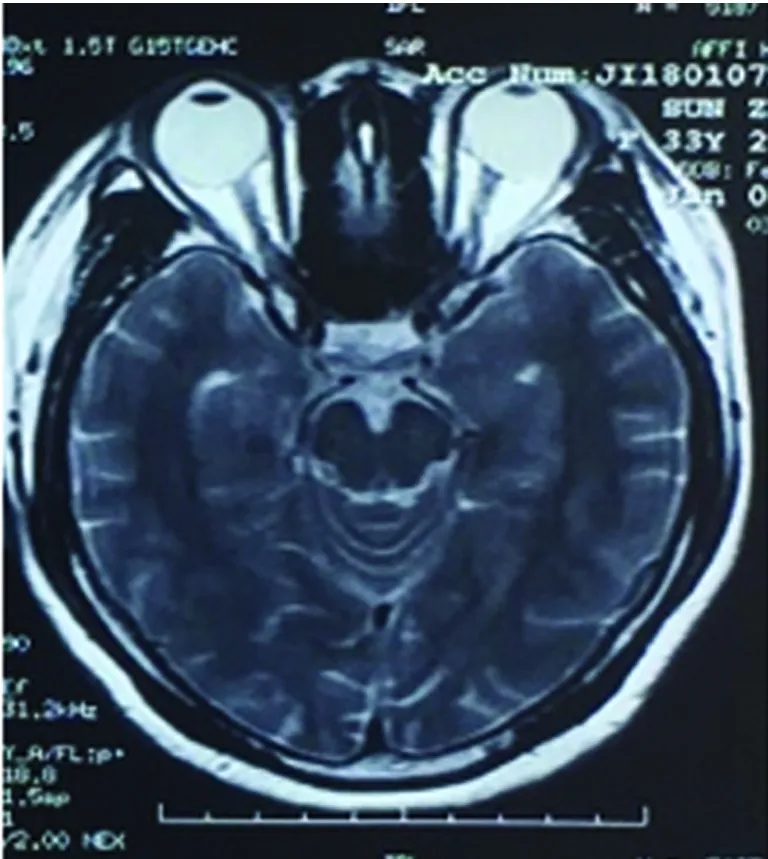
图2 头部MRI平扫左侧视神经未见明显增粗及高信号
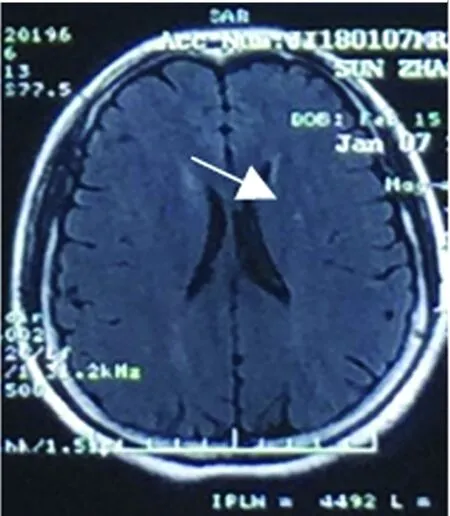
图3 头部MRI平扫可见左侧放射冠区点状脱髓鞘

图4 胸髓MRI可见T6-T7水平髓内异常信号
2 讨 论
视神经脊髓炎又称Devic病在1894年由Devic等提出,后来临床发现NMO存在缓解复发,很长一段时间归类于多发性硬化。直到NMO-IgG的发现[1],才将NMO视为一种独立的疾病。由于临床上常见到复发性视神经炎、复发性脊髓炎、伴有脑部病损或自身免疫病的视神经或脊髓炎,于是在2007年由Wingerchuk等[2]提出了NMOSD。此后又有些研究发现一些患者最初发病时仅有颅内典型部位病损及相关临床症状而无视神经炎或脊髓炎,但后续反复发作的病情最终满足视神经脊髓炎的诊断,因此在2015年提出了NMOSD[3]新的诊断标准。NMOSD的核心临床症状,包括视神经炎、急性脊髓炎、极后区综合征(发作性呃逆、恶心或呕吐,无法用其他原因解释)、急性脑干综合征、发作性嗜睡或急性间脑症状(伴MRI上NMOSD典型的间脑病损)、大脑综合征(伴NMOSD典型的大脑病损)。根据血液中AQP4抗体检出情况被分为AQP4抗体(+)和AQP4抗体(-)两组。最近有报道在 AQP4 抗体阴性的 NMOSD患者可检测到MOG抗体,主要表现为复发的视神经炎或长节段横贯性脊髓炎,其也见于横贯性脊髓炎或急性播散性脑脊髓炎[4]。少有人报道MOG 抗体及 AQP4 抗体双阳性的NMOSD病例。Sato等[4]检测215例NMOSD的血清MOG 抗体及 AQP4 抗体结果显示7.4%(16/215)患者MOG抗体阳性,64.7%(139/215)患者AQP4抗体阳性,27.9%(60/215)双抗体阴性,无1例患者双抗体阳性。Hoftberger 等[5]检测的174 例NMOSD患者血清,发现2例两种抗体均为阳性,且MOG抗体和 AQP4 抗体阳性者临床表现与AQP4抗体阳性者相似。
研究显示进入中枢神经系统的AQP4抗体与分布于星形胶质细胞的AQP4结合,激活补体系统,产生补体依赖性细胞毒性和细胞介导的抗体依赖性细胞毒性作用[6],最终导致星形胶质细胞损伤、轴索变性、神经元死亡。MOG抗体是一种存在于中枢神经系统(central nervous system,CNS)髓鞘最外层含量极微的髓鞘蛋白成分,一项研究将MOG 抗体注射到小鼠的大脑内,抗体仅会造成暂时性的髓鞘损伤及脱失,不会引起炎性细胞的浸润以及轴索的改变,而且这些改变可以在2 w内恢复[7]。因此许多研究显示与AQP4抗体阳性的NMOSD 患者相比MOG抗体阳性的NMOSD 总体预后较好,最后随访扩展残疾状态评分量表(EDSS)评分较 AQP4 抗体阳性者低[4,8~11]。研究还发现AQP4抗体阳性的NMOSD 好发于女性,病变脊髓节段更长(>3个椎体节段);易复发及伴有其他自身抗体[4,12]。而MOG抗体阳性者常见于男性及高加索人[5,9,10,13],脊髓MRI表现为炎性脱髓鞘病灶,脊髓病灶长度一般为2~5个椎体节段[14],且更易侵犯视神经。一项包含100例NMOSD患者研究发现:MOG抗体阳性的患者约有40%同时出现视神经炎与脊髓炎,并且明显高于AQP4抗体阳性组[10]。本患者女性,合并甲状腺功能异常,反复发作数次临床事件,每次激素治疗敏感,短期内恢复,未遗留明显后遗症,2次同侧视神经损害,并且出现短节段胸髓病灶。此患者的临床表现更类似于MOG抗体阳性的NMOSD患者。
本患者不仅AQP4抗体及MOG抗体阳性,还合并甲状腺功能异常,亚临床甲亢,甲状腺抗体检测阴性,且其姐姐患有甲亢,考虑患者自身免疫功能紊乱,可能多种机制参与了临床事件的发生。有研究显示 MOG 抗体阳性者可向 AQP4 抗体转化[11]。本患者前3次发病过程中均未进行检测MOG抗体及AQP4抗体,此次血液检查两抗体均阳性,不知本患者是否是转化中的1例。
MOG抗体及AQP4抗体所引发的NMOSD,发病机制不同,有着相似的临床表现,又各有侧重,患者的临床预后也有一定的差异,建议NMOSD患者检查时不仅完善AQP4抗体检查,也同时完善MOG抗体检查,以便利于判断患者的预后及治疗。
