新生儿甲基丙二酸血症的临床特点分析
2018-04-10吴海兰董世霄翁景文沈艳华
吴海兰,董世霄,刘 红,靳 绯,翁景文,沈艳华,姜 敏
(国家儿童医学中心,首都医科大学附属北京儿童医院新生儿中心NICU,北京 100045;*通讯作者,E-mail:dongsx.hi@163.com)
甲基丙二酸血症(methylmalonic acidemia,MMA)也称甲基丙二酸尿症(methylmalonic aciduria,MMA),是由于甲基丙二酰辅酶A变位酶(methylmalonyl-CoA mutase,MCM)活性低下或其辅酶钴胺素(维生素B12)代谢缺陷所致。这是先天性有机酸代谢障碍中较常见的一种常染色体隐性遗传病,迄今已发现7种亚型。患儿临床表现复杂,国内资料显示,约半数的患儿在新生儿期以非特异性症状起病[1],临床诊断困难,症状类似缺血缺氧性脑病、败血症、贫血等疾病[2,3]。MMA患儿可以三系减低或两系减低为突出表现,易引起误诊,延误治疗。为此,我们对2008-2016年我院NICU诊治的21例甲基丙二酸血症患儿进行了回顾性研究。
1 资料和方法
1.1 一般资料
2008-2016年我院NICU收治甲基丙二酸血症患儿21例,男15例,女6例,其中合并同型半胱氨酸血症6例(28.6%)。
1.2 诊断方法
采用气相色谱质谱联用分析(gas chromatography-mass spectrometry,GC/MS)检测尿液中甲基丙二酸及其代谢产物水平,尿液中甲基丙二酸浓度高于参考值(0.2-3.6 mmol/mol肌酐)100倍以上,并除外因叶酸、维生素B12缺乏等引起的继发性MMA则诊断遗传性MMA[4]。通过液相色谱串联质谱技术(liquid chromatography tandem mass spectrometry,LC·MS)进行血液氨基酸、酯酰肉碱谱分析,并检测血清同型半胱氨酸浓度(正常参考值1.7-17.4 μmol/L),单独型甲基丙二酸血症患儿血清同型半胱氨酸正常,合并同型半胱氨酸血症的患儿血清同型半胱氨酸显著增高。对部分上述检测异常的患儿进一步送检基因分型。
1.3 研究方法
1.3.1资料收集回顾性分析21例患儿的临床资料,包括:性别、发病年龄、确诊时间、家族史、发病时临床表现、治疗经过、辅助检查等。
1.3.2病例随访对确诊患儿每半年进行电话或门诊随访至2岁。
2 结果
2.1 发病情况及临床表现
21例中男15例,女6例。发病年龄生后5 h-24 d,确诊日龄7-31 d(见表1)。最多见的临床表现依次为喂养困难(16例)、反应弱(13例)、惊厥(7例)、发热(3例)。3例有同胞不明原因死亡家族史。初步诊断仅有3例被考虑为先天性遗传代谢病。
表121例MMA患儿的发病情况及临床表现
Table1Clinicalcharacteristicsof21casesofMMA
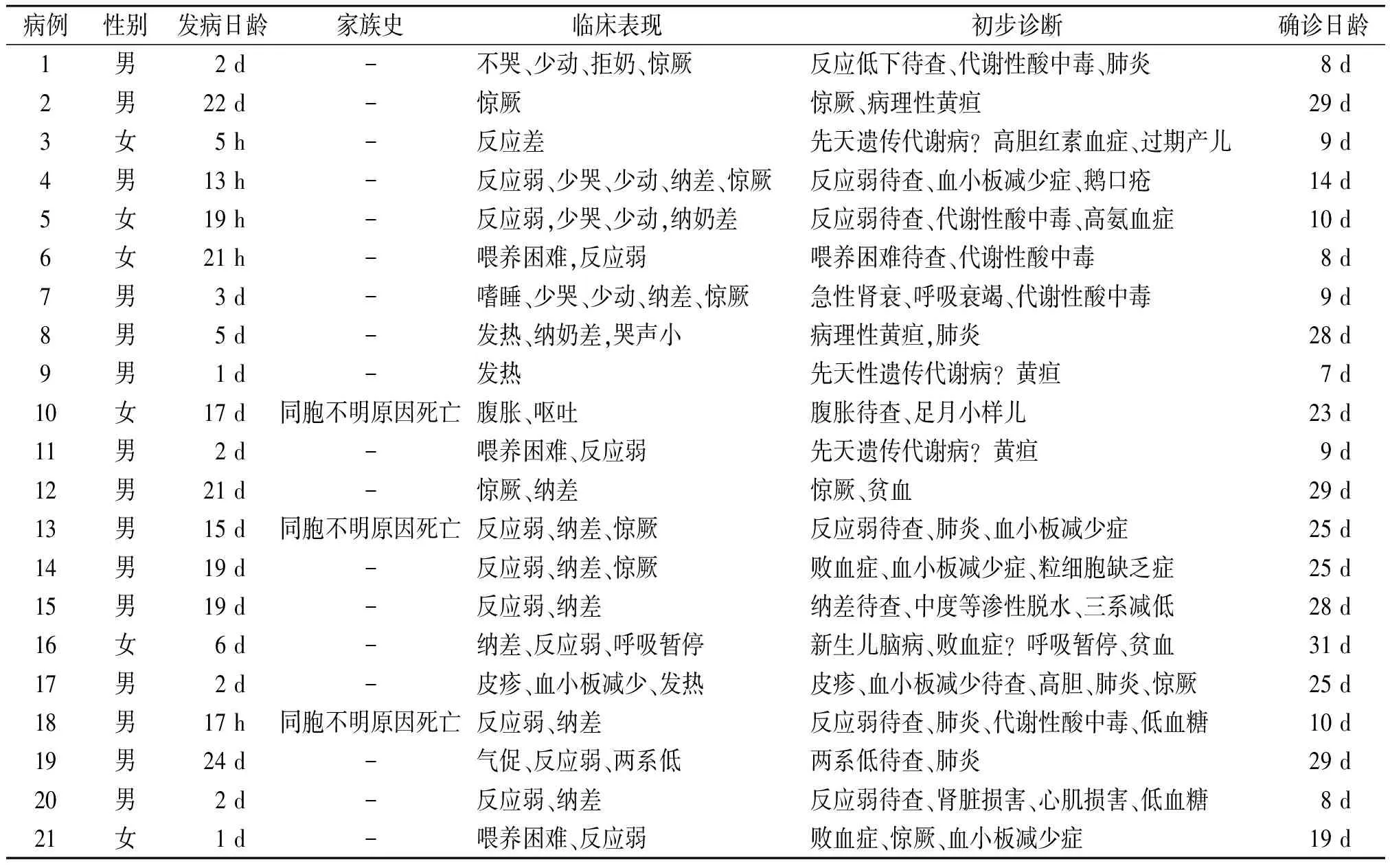
病例性别发病日龄家族史临床表现 初步诊断 确诊日龄1男2d-不哭、少动、拒奶、惊厥反应低下待查、代谢性酸中毒、肺炎8d2男22d-惊厥惊厥、病理性黄疸29d3女5h-反应差先天遗传代谢病?高胆红素血症、过期产儿9d4男13h-反应弱、少哭、少动、纳差、惊厥反应弱待查、血小板减少症、鹅口疮14d5女19h-反应弱,少哭、少动,纳奶差反应弱待查、代谢性酸中毒、高氨血症10d6女21h-喂养困难,反应弱喂养困难待查、代谢性酸中毒8d7男3d-嗜睡、少哭、少动、纳差、惊厥急性肾衰、呼吸衰竭、代谢性酸中毒9d8男5d-发热、纳奶差,哭声小病理性黄疸,肺炎28d9男1d-发热先天性遗传代谢病?黄疸7d10女17d同胞不明原因死亡腹胀、呕吐腹胀待查、足月小样儿23d11男2d-喂养困难、反应弱先天遗传代谢病?黄疸9d12男21d-惊厥、纳差惊厥、贫血29d13男15d同胞不明原因死亡反应弱、纳差、惊厥反应弱待查、肺炎、血小板减少症25d14男19d-反应弱、纳差、惊厥败血症、血小板减少症、粒细胞缺乏症25d15男19d-反应弱、纳差纳差待查、中度等渗性脱水、三系减低28d16女6d-纳差、反应弱、呼吸暂停新生儿脑病、败血症?呼吸暂停、贫血31d17男2d-皮疹、血小板减少、发热皮疹、血小板减少待查、高胆、肺炎、惊厥25d18男17h同胞不明原因死亡反应弱、纳差反应弱待查、肺炎、代谢性酸中毒、低血糖10d19男24d-气促、反应弱、两系低两系低待查、肺炎29d20男2d-反应弱、纳差反应弱待查、肾脏损害、心肌损害、低血糖8d21女1d-喂养困难、反应弱败血症、惊厥、血小板减少症19d
2.2 实验室检查
21例MMA患儿中,高氨血症11例(52.4%)、代谢性酸中毒10例(47.6%)、大细胞贫血8例(38.1%)、粒细胞减低7例(33.3%)、血小板减少7例(33.3%)、肝功能异常3例(14.3%)。8例行血同型半胱氨酸测定,显著增高6例(见表2)。
表221例MMA患儿的实验室检查
Table2Laboratoryresultsof21casesofMMA
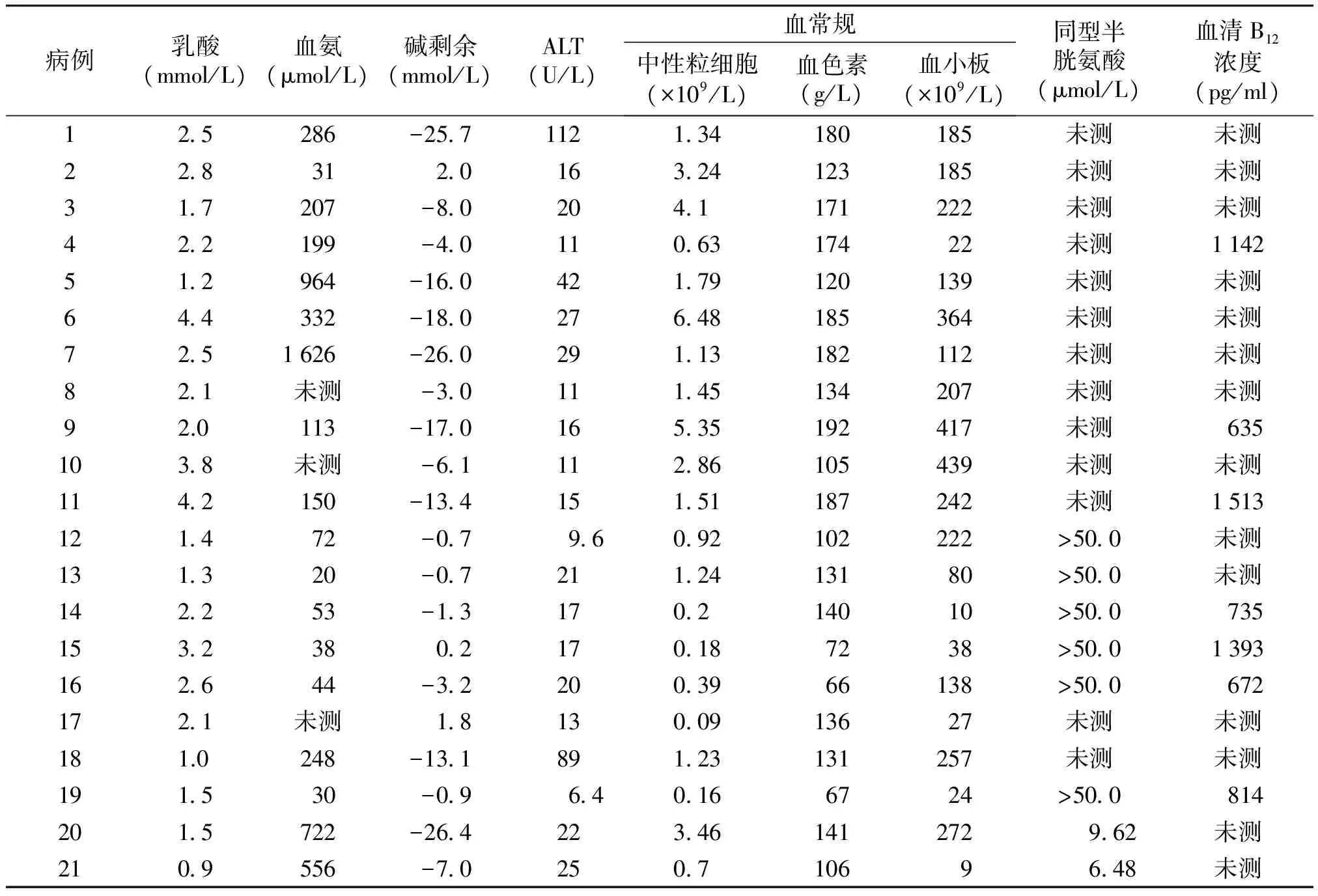
病例乳酸(mmol/L)血氨(μmol/L)碱剩余(mmol/L)ALT(U/L)血常规中性粒细胞(×109/L)血色素(g/L)血小板(×109/L)同型半胱氨酸(μmol/L)血清B12浓度(pg/ml)125286-257112134180185未测未测228312016324123185未测未测317207-802041171222未测未测422199-401106317422未测1142512964-16042179120139未测未测644332-18027648185364未测未测7251626-26029113182112未测未测821未测-3011145134207未测未测92.0113-17016535192417未测 6351038未测-6111286105439未测未测1142150-13415151187242未测1513121472-0796092102222>500未测131320-072112413180>500未测142253-13170214010>500 73515323802170187238>5001393162644-322003966138>500 6721721未测181300913627未测未测181.0248-13189123131257未测未测191530-09640166724>500 8142015722-26422346141272 962未测2109556-7025071069 648未测
参考值范围:乳酸0.5-2 mmol/L;血氨18-72 μmol/L;碱剩余-3~3 mmol/L;ALT 5-40 U/L;中性粒细胞(0.72-4.6)×109/L;血色素110-160 g/L;血小板(100-400)×109/L;同型半胱氨酸1.7-17.4 μmol/L;血清B12浓度140-960 pg/ml
2.3 头颅影像学检查
MMA的颅脑影像学表现非特异。21例中12例行头颅CT检查,均表现为双侧大脑半球脑白质密度对称性减低;5例行头部MR检查,提示额顶枕叶皮层下脑白质呈长T1长T2信号,类似缺氧缺血性脑病样改变。
2.4 诊断经过
21例患儿经尿液GC/MS分析发现甲基丙二酸水平明显升高,8例进行血同型半胱氨酸测定,发现合并同型半胱氨酸血症6例(占总例数的28.6%),其中2例甲基丙二酸血症合并同型半胱氨酸血症患儿接受了基因检测,均为CblC型。
2.5 治疗及随访
确诊甲基丙二酸血症后,对于单独型MMA患儿,给予大剂量维生素B121-2 mg/次、1次/d肌内注射,3-7 d后改为维生素B12l mg/d每周1-2次肌内注射或口服甲钴胺1 mg/d,左卡尼汀[30-200 mg/(kg·d)]静脉输注或口服,低蛋白饮食[将天然蛋白质控制在0.5-1.5 g/(kg·d)],并给予祛除蛋氨酸、缬氨酸、苏氨酸的特殊配方奶粉。对于合并型MMA患儿,给予维生素B121-2 mg/次、1次/d肌内注射,3-7 d后改为每周1-2次肌内注射维生素B121 mg或口服甲钴胺1 mg/d,左卡尼汀30-200 mg/(kg·d)静脉输注或口服,甜菜碱100-500 mg/(kg·d)口服,正常饮食。
21例患儿中7例失访,4例放弃治疗后死亡,2例于1-2岁时死亡,死亡率28.6%;7例患儿经过治疗后临床症状改善,但遗留轻-重度智力体力发育障碍,致残率33.3%;1例随访至2岁生长发育基本正常。
3 讨论
MMA是我国先天性有机酸代谢障碍中较常见的一种常染色体隐性遗传病,病因是甲基丙二酰辅酶A变位酶(methylmalonyl-CoA mutase,MCM)活性低下或其辅酶钴胺素(维生素B12)代谢缺陷所引起[5]。各国发病率不同,约为1/50 000-1/100 000[6]。根据酶缺陷类型的不同将MMA分为7种类型,其中MCM缺陷包括MCM完全缺乏(mut0)和部分缺乏(mut-)2种类型;钴胺素代谢障碍包括CblA、CblB、CblC、CblD、CblF 5种类型。mut0、mut-、CblA和CblB型表现为单纯型MMA,CblC、CblD和CblF型则表现为MMA伴同型半胱氨酸血症(简称合并型MMA)。尿GC/MS分析和血MS/MS分析是确诊MMA的首选且较快捷的诊断方法。基因突变分析是确诊MMA及进行诊断分型的最可靠依据。
新生儿期MMA的临床表现缺乏特异性,误诊率较高。主要表现为反应弱、喂养困难、呕吐、代谢性酸中毒、惊厥、意识障碍等,类似新生儿缺氧缺血性脑病、败血症等常见疾病,常合并肺炎等感染性疾病及多系统损害,容易被误诊,死亡率、致残率很高[7,8]。本组21例甲基丙二酸血症患儿起病表现为多种异常,以反应弱、喂养困难、惊厥最多见,实验室检查提示血氨增高11例、其中10例合并代谢性酸中毒。发病后可很快进展为呼吸衰竭、昏迷等代谢危象状态。建议对于新生儿期有上述表现的患儿及早进行尿液和血液的代谢筛查,争取早期诊断、正确治疗、改善预后。合并型MMA是MMA中较常见的类型,其中CblC亚型是我国MMA伴同型半胱氨酸血症中最常见的类型[9]。甲基丙二酸及其代谢产物可导致患儿神经细胞凋亡、脱髓鞘样改变,引起生长发育障碍、神经系统异常[10],并可导致肝、肾、胃肠、皮肤等多脏器损害。同型半胱氨酸为含巯基氨基酸,是胱硫醚和蛋氨酸转硫化和甲基化代谢旁路中形成的中间体,同型半胱氨酸蓄积可导致血管内皮损伤和血液系统异常如血栓性血小板减少、溶血尿毒综合征[11]、骨髓抑制[12]等。由于合并型MMA兼有甲基丙二酸及同型半胱氨酸的毒性作用,部分还伴有低蛋氨酸血症,因此合并型患儿的临床表现更为复杂多样[13]。常规实验室检查中代谢性酸中毒及高血氨较少见,多提示血液系统损伤[14]。本组中7例以三系或两系血细胞减低为突出表现,入院诊断为血小板减少症、粒细胞减少,伴或不伴贫血,容易误诊为败血症、血小板减少性紫癜等,误诊率高。本组中检测出的6例合并型MMA患儿均不合并高氨血症及代谢性酸中毒。故新生儿期以三系减低或两系减低为突出表现的患儿,即使不存在血氨增高及代谢性酸中毒,仍建议及早进行尿筛查及血串联质谱检测,及时明确诊断。
中国MMA大多为MMA伴同型半胱氨酸血症,约占60%-80%[15],而CblC亚型是其中较常见的类型。本组患儿中8例进行了血同型半胱氨酸检测,其中6例检测出血同型半胱氨酸浓度大于50 μmol/L,诊断甲基丙二酸血症合并同型半胱氨酸血症,约占总例数的28.6%,其中基因检测2例,均为CblC型。与报道不一致,原因主要是由于早期对本病认识不够,未能完善血同型半胱氨酸水平测定及基因检测,但6例高同型半胱氨酸血症,占8例送检血同型半胱氨酸浓度测定的75%,与文献报道相符。甲基丙二酸血症是否合并同型半胱氨酸血症,治疗方法不同,单纯型甲基丙二酸血症饮食治疗原则是限制天然蛋白质,补充去除蛋氨酸、缬氨酸、苏氨酸、丝氨酸的特殊奶粉或氨基酸粉,而甲基丙二酸血症合并同型半胱氨酸血症的患儿自身蛋氨酸合成障碍,特殊饮食治疗将进一步加重蛋氨酸缺乏、医源性低甲硫氨酸血症,常引起肢端皮炎样皮疹、营养不良等一系列损害[16,17]。因此,针对单独型甲基丙二酸血症患儿的特殊配方奶不适于合并同型半胱氨酸血症的患儿,此类患儿在补充维生素B12、叶酸、左旋肉碱、甜菜碱的基础上给予天然饮食,更利于疾病控制与营养发育[18]。建议对于甲基丙二酸血症患者及早进行血清或尿液同型半胱氨酸测定,并完善血清叶酸或维生素B12的检测(尤其合并贫血的患儿),以除外继发性MMA及营养性巨幼细胞性贫血[6]。争取早期明确诊断,合理治疗,以改善患儿的长期预后。
MMA的预后主要取决于分型和诊断治疗的时间。早发型(1岁以内发病的)甲基丙二酸血症合并同型半胱氨酸血症患儿起病凶险、死亡率很高,晚发型患儿预后相对较好[7]。变位酶缺陷(mut0型)导致的单纯型甲基丙二酸血症患儿病情严重,起病急,生后数小时至几天内发病,新生儿期死亡率很高,预后极差[8]。新生儿期发病的患儿均为早发型MMA,多数预后不良,死亡率、致残率较高。本组中除7例失访外,仅1例生长发育基本正常,6例死亡(28.6%),7例留有不同程度生长发育障碍(33.3%)。随着对本病认识的不断提高,希望越来越多的患儿得以及时的诊断和治疗,尽可能地改善预后。
参考文献:
[1]李薇,刘芳.新生儿甲基丙二酸血症六例[J].中国综合临床,2016,32(12):1128-1129.
[2]Yang Y, Sun F, Song J,etal. Clinical and biochemical studies on Chinese patients with methylmalonic aciduria[J]. J Child Neurol, 2006, 21(12):1020-1024.
[3]Fowler B, Leonard JV, Baumgartner MR. Causes of and diagnostic approach to methylmalonic acidurias[J]. J Inherit Metab Dis, 2008, 31(3):350-360.
[4]Hori D, Hasegawa Y, Kimura M,etal. Clinical onset and prognosis of Asian children with organic acidemias, as detected by analysis of urinary organic acids using GC/MS, instead of mass screening[J]. Brain Dev Jpn, 2005, 27(1):39-45.
[5]Deodato F, Boenzi S, Santorelli FM,etal. Methylmalonic and propionic aciduria[J]. Am J Med Genet C, 2006, 142C:104-112.
[6]臧文涛,盛光耀,王璐,等.以大细胞性贫血为突出表现的甲基丙二酸血症1例[J].中国当代儿科杂志,2015,17(7):755-756.
[7]张尧,宋金青,刘平,等.甲基丙二酸尿症合并同型半胱氨酸血症57例临床分析[J].中华儿科杂志,2007,45(7):513-517.
[8]Martinelli D, Dotta A, Massella L,etal. Cobalamin C defect presenting as severe neonatal hyperammonemia[J]. Eur J Pediatr, 2011, 170(7):887-890.
[9]Liu MY, Yang YL, Chang YC,etal. Mutation spectrum of MMACHC in Chinese patients with combined methylmalonic aciduria and homocystinuria[J]. J Hum Genet,2010,55(9):621-626.
[10]Tao-Yun JI, Zhang YH, Fei-Tian LI,etal. Peripheral nervous impairment in a patient with methylmalonic aciduria combined with hyperhomocysteinemia[J]. J Peking Univ, 2013,45(2):303-306.
[11]Menni F, Testa S, Guez S,etal. Neonatal atypical hemolytic uremic syndrome due to methylmalonic aciduria and homocystinuria[J]. Pediatr Nephrol, 2012, 27(8):1401-1405.
[12]Watkins D, Rosenblatt DS. Inborn errors of cobalamin absorption and metabolism[J]. Am J Med Genet C, 2011, 157C(1):33-44.
[13]Martinelli D, Deodato F, Dionisi-Vici C. Cobalamin C defect: natural history, pathophysiology, and treatment[J]. J Inherit Metab Dis, 2011, 34(1):127-135.
[14]黄倬,韩连书,叶军,等.甲基丙二酸血症患者143例资料分析[J].中华内分泌代谢杂志,2014,30(6):490-494.
[15]刘玉鹏,马艳艳,吴桐菲,等.早发型甲基丙二酸尿症160例新生儿期异常表现[J].中华儿科杂志,2012,50(6):410-414.
[16]赵英,张月华,杨艳玲,等.甲基丙二酸血症合并癫痫27例临床特点及预后分析[J].中国实用儿科杂志,2011,26(1):37-40.
[17]宇亚芬,黎芳,麻宏伟.cbIC型甲基丙二酸血症基因型与临床表型及疗效的关系[J].中国当代儿科杂志,2015,17(8):769-774.
[18]Carrillocarrasco N, Chandler RJ, Venditti CP. Combined methylmalonic acidemia and homocystinuria, cblC type.I. Clinical presentations, diagnosis and management[J]. J Inherit Metab Dis, 2012, 35(1):91-102.
