假性甲状旁腺功能减退症合并低钾血症临床特点分析
2017-09-12王玉成周红文陈欢欢袁庆新李剑波
张 维,张 梅,王玉成,崔 岱,周红文,陈欢欢,袁庆新, 李剑波,杨 涛
(1.南京医科大学第一附属医院 内分泌科,江苏 南京 210029; 2.芜湖市第二人民医院 内分泌科,安徽 芜湖 241000)
·论著·
假性甲状旁腺功能减退症合并低钾血症临床特点分析
张 维1,2,张 梅1,王玉成1,崔 岱1,周红文1,陈欢欢1,袁庆新1, 李剑波1,杨 涛1
(1.南京医科大学第一附属医院 内分泌科,江苏 南京 210029; 2.芜湖市第二人民医院 内分泌科,安徽 芜湖 241000)
目的 分析假性甲状旁腺功能减退症(pseudohypoparathyroidism,PHP)合并低钾血症患者的临床特征。方法 回顾性分析2006年7月至2016年4月期间在我院住院治疗的13例PHP患者的临床资料。结果 13例患者中男5例,女8例,均无典型Albright遗传性骨营养不良症(Albright’s hereditary osteodystrophy,AHO)躯体畸形,临床表现84.6%的患者有手足抽搐症状,38.5%的患者有四肢乏力、麻木症状;实验室检查6例合并低血钾,10例合并甲状腺功能异常;合并低血钾组病程较长,血钾在3.0~3.5 mmol/L,其他临床表现及生化指标与非低血钾组相比差异无统计学意义。结论 PHP患者易合并低钾血症及亚临床甲状腺功能减低。
假性甲状旁腺功能减退症;低钾血症; 甲状腺功能减退症
假性甲状旁腺功能减退症(pseudohypoparathyroidism,PHP)是一种少见的多基因遗传性疾病,其发病基因可在X染色体上,也可在常染色体上,表现为显性或隐性遗传,男女发病率约为1∶2。该疾病临床表现多样,共同特征为:有低血钙、高血磷,血清甲状旁腺素(PTH)水平升高,靶组织对活性PTH无反应,部分患者还伴有特殊的躯体畸形。PHP为临床少见疾病,对其中合并低钾血症者的报道及研究更少。本文回顾分析了住院治疗的PHP合并低钾血症患者的临床资料,并对其特点进行了总结,以期提高对PHP合并低钾血症的认识。
1 资料与方法
1.1 病例选择 2006年7月至2016年4月南京医科大学第一附属医院内分泌科确诊为PHP患者13例。PHP主要诊断依据[1]:①有甲状旁腺功能减退症的生化改变(低血钙、高血磷)。②靶组织对活性PTH无反应。③血PTH增高(要排除肾脏功能不全),碱性磷酸酶正常。④多数患者还伴有特殊的躯体畸形。
1.2 方法 回顾性分析研究。① 病史及体格检查:包括性别、年龄、发病年龄、病程、手足搐搦、癫痫样发作、其他主要不适、Albright遗传性骨营养不良症(Albright’s hereditary osteodystrophy,AHO)表型。 AHO表型包括:脸圆、身材矮小、肥胖、掌(跖)骨短、皮下骨性结节。② 实验室检查:血钾、血钙、血磷、血钠、血氯、血脂、肝肾功能、血PTH、甲状腺功能,24小时尿电解质。③ 影像学检查:头颅CT、全身多处骨平片、甲状旁腺超声或核素显像。所有患者根据血钾水平,分为低血钾组(血钾<3.5 mmol/L),非低血钾组(血钾≥3.5 mmol/L)。

2 结 果
2.1 一般临床特征 ①发病年龄:发病年龄2~44岁,平均年龄26.5岁,发病高峰年龄在11~30岁(9例),10岁前(2例)及30岁(2例)。② 性别:13例PHP患者中女8例,男5例。③病程:患者就诊时病程1~20年不等,平均病程7.1年。④临床表现:84.6%的患者有手足抽搐症状,且可为唯一症状,38.5%的患者有四肢乏力、麻木症状,本文13例患者均未见有典型癫痫样发作症状。13例患者均无典型AHO体征(小圆脸、矮身材、掌指骨粗短等)。
2.2 主要实验室检查 ①所有患者均有低血钙(1.66±0.25 mmol/L,正常值2.20~2.65 mmol/L)、高血磷(1.93±0.63 mmol/L,正常值0.81~1.45 mmol/L)、血PTH增高(455.88±275.57 pg/ml,正常值12.0~88.0 pg/ml)。② 13例患者中有10例合并甲状腺功能减退(甲减)(76.9%),其中7例为亚临床甲减,3例为轻度临床甲减。③其他辅助检查:全身骨骼平片提示异常2例,头颅CT提示多发颅内钙化灶6例,影像学检查提示甲状旁腺增生3例。
2.3 伴或不伴低血钾的PHP患者临床特征比较 ①低血钾发生情况:13例患者中有6例合并低钾血症,均为轻度低血钾,血钾在3.0~3.5 mmol/L。低血钾组有5例患者进行了24小时尿电解质测定,非低血钾组有4例患者进行了尿电解质测定,两组比较差异无统计学意义,见表1。② 低血钾组与非低血钾组临床特点比较:低血钾组病程较非低血钾组长(P=0.05),并且患者麻木、乏力症状较非低血钾组多见(66.7% vs 14.3%,P=0.086)。两组间性别、年龄无明显差异。甲减发生率、骨骼平片及头颅CT阳性率差异无统计学意义,见表2。③ 低血钾组与非低血钾组实验室检测结果比较:低血钾组与非低血钾组的血钙、血磷、血PTH水平差异均无统计学意义(P>0.05),见表3。
2.4 随访 6例合并低钾血症的患者,给予补充活性维生素D及钙剂的治疗,3个月后随访,复查血钾、血钙均恢复正常。
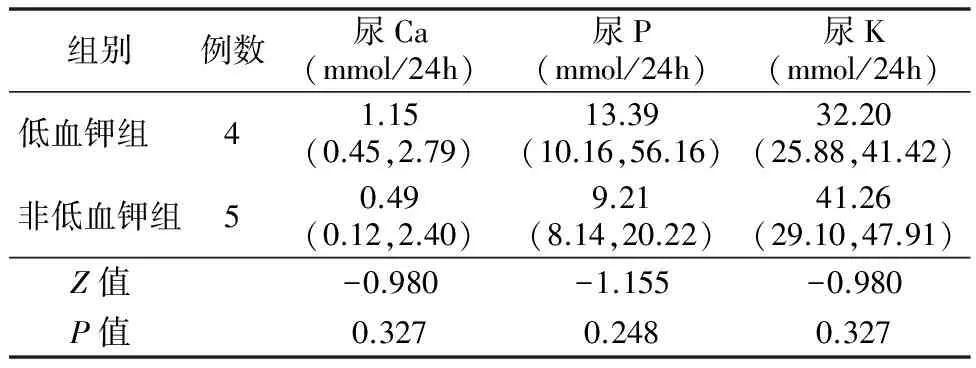
表1 2组尿电解质比较[中位数(25%,75%)]
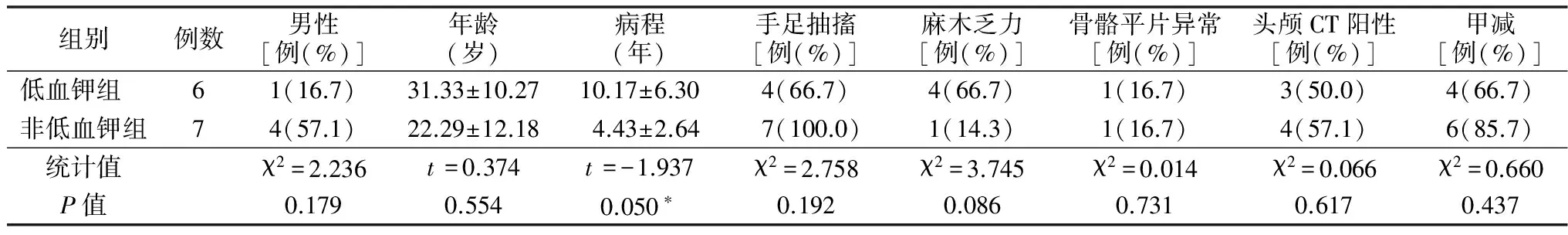
表2 2组临床特点比较
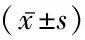
表3 2组实验室指标比较
3 讨 论
PHP是一种遗传性甲状旁腺疾病,由Albright等[2]于1942年首次报道,1979年 Aurbach统计全世界约100例该病患者。PHP主要因为靶器官(肾脏、骨骼等的细胞膜G蛋白耦联受体)对PTH的抵抗,从而使PTH分泌代偿性增加。临床上以低钙性手足抽搐、高血磷、PTH抵抗(包括维生素D生成减少)、AHO(身材矮小、肥胖、圆脸、短指趾畸形,有时伴有智力减退)为主要特征[3]。该疾病在临床上并不多见,因其多有手足抽搐、智力低下等症状,易被误诊为癫痫等其他疾病[4-6]。
PTH抵抗是各种类型的PHP中最主要的生物学异常,由于近端肾小管中Gsα表达的减少或缺乏,导致严重的低钙血症,同时也致1,25(OH)维生素D合成减少。此外,异常的PTH下调了钠依赖的磷酸盐共转运体(NPT2a)和NPT2c的水平,从而导致尿磷排出减少,血磷升高[7-8]。PHP典型的生化表现为低血钙、高血磷、高PTH血症,同时血镁水平需在正常范围。因为轻度的镁缺乏会抑制PTH活性,重度的镁缺乏会抑制PTH的分泌,故正常血镁水平也是PHP诊断成立的要素之一[9]。本文总结13例病例发现,PHP合并低钾血症6例(46.2%),均为轻度低血钾,血钾在3.0~3.5 mmol/L,且低血钾组24小时尿钾水平与非低血钾组相比,差异无统计学意义。PHP合并低钾血症国内外报道均较少,王转锁等[10]总结20例PHP患者临床特征发现有20%患者合并低钾血症,其中有2例诊断为Bartter综合征。Iba等[11]曾报道1例PHP女性,合并Bartter综合征,应用小剂量活性维生素D(0.5 μg/d)及安体舒通(50 mg/d)治疗,即可维持血钙、血钾在正常范围。本文报道的6例合并低钾血症患者,有1例同时合并低镁、代谢性碱中毒、血醛固酮、肾素水平增高,临床诊断为PHP合并Gitelman综合征,但该病例未经基因诊断证实。另外5例患者排除其他原因导致的低钾血症,给予补充活性维生素D及钙剂后,血钙、血钾均恢复正常。临床上可导致低钾血症的原因很多,根据病因可以分为钾摄入减少、钾向细胞内转移、肠道以及肾脏失钾。分布于人体肌肉组织细胞膜上的钠-钾泵保证了人体98%的钾分布在细胞内,只有2%的钾在细胞外,因而即使很微量的钾离子在细胞内外的转移,也会导致血钾的较大波动。已知的胰岛素、β肾上腺素能受体激动剂、碱中毒,以及醛固酮均能激活钠-钾ATP泵,促使钾向细胞内转移[12]。PTH是否也有类似的作用并无确切研究。肾脏是调节血钾平衡的最主要的器官,几乎所有滤过的钾都会在近端肾单位被重吸收,分析PHP导致低血钾的机制为肾性失钾可能较大:① 体内的钾离子通道有多种类型,在肾脏髓袢升支粗段,可分为管腔膜钾离子通道和管周膜钾离子通道两种。其中管腔膜钾离子通道对钾离子的再循环起重要作用,CAMP-PKA途径对部分钾离子通道的活性起到促进作用。PHP的患者,因肾脏PTH抵抗,CAMP的形成减少或CAMP作用缺陷,可能导致管腔膜钾离子通道活性受抑制,使得髓袢升支粗段的钾离子再循环障碍,管腔内K+浓度减少,抑制Na+-K+-2Cl-同向共转运体的活性,使得钠离子丢失,远端肾小管钠重吸收增加,与氢、钾交换使得钾的分泌增多。这与Bartter综合征导致低钾血症的作用机制有类似之处[13]。②长期低血钙可能导致肾小管上皮细胞变性,继发功能障碍也可能导致钾的重吸收障碍,从而引起低血钾。
本文中总结的13例患者有10例存在轻度甲减,其中大部分为亚临床甲减,仅有促甲状腺激素(TSH)轻度增高,考虑为激素抵抗所致。PHP可以分为Ⅰ型(Ia、Ib、Ic)和Ⅱ型,Ⅰ型的特征性病变为编码异三聚体G蛋白的α亚单位(Gsα)的GNAS基因的异常,Gsα在大多数细胞中都是双等位基因翻译,但在一些组织中来自母系的等位基因占优势(比如近端肾小管、甲状腺、卵巢G细胞、性腺细胞等)[14]。Ia型临床上最常见,多存在特有的发育和体态异常,称为AHO,常可伴其他激素抵抗,如TSH、胰高血糖素、促性腺激素等,继而出现相应器官功能的低下,主要原因为GNAS基因外显子区域来自母系的等位基因存在混杂突变所致;Ib型大多不伴有AHO的临床表现,其病因主要为GNAS基因部分区域的甲基化异常,导致来自母系的等位基因的翻译减少。无论是Ia型还是Ib型,都可能存在PTH和TSH抵抗现象。Molinaro等[15]对4例通过染色体检查确诊为Ib型PHP的0.5~15岁的患者研究发现,亚临床甲减可为其首发表现,且可于其他临床症状出现前3~20年出现。PHPIb的不同表型可能与GNAS基因的特征性表观遗传学改变有关。其中一些常染色体显性表型与来自母体的GNAS外显子NESP和(或)AS3-4的缺失有关[16],例如:GNAS外显子A/B、AS或XL的甲基化缺失,可导致患者出现不同的临床表型。最常见的缺陷是编码突触融合蛋白16的STX16基因的位点缺失。大部分的PHPIb患者都是散发的,其中只有一小部分被证实是来自父系的染色体20q单系单倍体型或单倍异二体型所致,更多的散发者并不能在分子水平得到证实[17-18]。所有这些患者的共同特征是来自母系的GNAS基因组甲基化改变,即丢失了所有来自母体的甲基化印迹,以及外显子NESP的双等位基因甲基化缺失[19-20]。推测可能是某种反式作用因子的缺乏所致[21-22]。Ic型可以看做是Ia型的变异,患者有典型的AHO表现,但血钙、血磷正常,血PTH增高,并无Ia型特征性的基因改变。本文总结的13例患者大多无典型的体态异常,大多数患者均合并TSH抵抗,从临床特征分析为Ib型可能较大,通过临床表现和生化检查很难将各亚型患者完全区分开,确诊需依赖染色体检查。因本文为回顾性分析,大部分患者均未行染色体检查,因实验室条件限制,也无法行PTH兴奋试验及Gsα测定等检查进一步明确分型,可在随访观察时予以完善。
综合上述分析,PHP患者合并低钾血症及TSH抵抗均较既往报道中常见,合并低钾血症者病程均较长,且多表现为轻度低血钾,尿钾排泄也无明显增加,因此在临床特征和辅助检查上与其他PHP患者较难区分。治疗上与普通PHP患者也无区别。临床遇到亚临床甲减及低钾血症患者时,需考虑合并本病可能,除常规检查外,需进一步测定血电解质、血PTH、24小时尿电解质、血浆醛固酮、肾素水平,以利于鉴别诊断。必要时可完善染色体检查确定诊断。
[1] 陈家伦.假性甲状旁腺功能减退症和假假性甲状旁腺功能减退症[M]/临床内分泌学.上海:科学技术出版社,2011:1374-1378.
[2] Ringel MD,Schwindinger WF,Levine MA,et al.Clinical implications of genetic defects in G proteins. The molecular basis of McCune-Albright syndrome and Albright hereditary osteodystrophy[J]. Medicine (Baltimore),1996,75(4) 171-184.
[3] Lemos MC, Thakker RV. GNAS mutations in Pseudohypoparathyroidism type 1a and related disorders[J].Hum Mutat, 2015,36(1):1l-19.
[4] 王莹,刘俊茹,高峰,等.假性甲状旁腺功能减退症2例报道[J].临床荟萃,2007,22(14):1050.
[5] 孙清元,王晓月,罗长青.假性甲状旁腺功能减退症误诊为癫痫3例[J].临床荟萃,2007,22(10):732-733.
[6] 韩凤山,吴建国.假性甲状旁腺机能减退症一例报告[J].世界最新医学信息文摘,2015,15(5):166-168.
[7] Mantovani G.Clinicalreview:pseudohypoparathyroidism:diagnosis and treatment[J].J Clin Endocrinol Metab,2011,96(10):3020-3030.
[8] Bastepe M, Turan S, He Q. Heterotrimeric G proteins in the control of parathyroid hormone actions[J]. J Mol Endocrinol, 2017,58(4): 203-224.
[9] Levine MA.An update on the clinical and molecular characteristics of pseudohypoparathyroidism[J].Curr Opin Endocrinol Diabetes Obes,2012,19(6): 443-451.
[10] 王转锁,任艳,田浩明.20 例假性甲状旁腺功能减退症临床分析[J].四川大学学报:医学版,2011,42(1):139- 140.
[11] Iba K, Morii H, Wada M,et al.A case report of pseudohypo-parathyroidism (Drezner'stypeI) associated with probable Bartter'ssyndrome[J].Endocrinoljpn,1981,28(5):595-604.
[12] Reid A, Jones G, Isles C.Hypokalaemiacommon things occur commonly-a retrospective survey[J]. JRSM Short Rep,2012,3(11):80.
[13] Weinstein AM.Amathematical modelof rat ascendingHenlelimb.Ⅲ.Tubularfunction[J].AM J Physiol Renal Physiol,2010,298(3):F543-556.
[14] Lemos MC, Thakker RV.Thankker. GNAS mutations in pseudohypoparathyroidism type 1a and related disorders[J]. Hum Mutat ,2015,36(1):11-19.
[15] Molinaro A, Tiosano D, Takatani R,et al.TSH elevations as the first laboratory evidence for pseudohypoparathyroidism type Ib (PHP-Ib) [J]. J Bone Miner Res, 2015,30(5):906-912.
[16] Rochtus A, Martin-Trujillo A, Izzi B,et al.Genome-wide DNA methylation analysis of pseudohypoparathyroidism patients with GNAS imprinting defects[J]. Clin Epigenetics,2016,8(1):1-12.
[17] Richard N,Abeguile G,Coudray N,et al.A new deletion ablating NESP55 causes loss of maternal imprint of A/B GNAS and autosomal dominant pseudohypoparathyroidism type Ib[J].J Clin Endocrinol Metab,2012,97(5):E863-867.
[18] Turan S,Ignatius J,Moilanen JS,et al.De novo STX16 deletions:an infrequent cause of pseudohypoparathyroidiism type Ib that should be excluded in sporadic Cases[J].J Clin Endocirnol Metab,2012,97(12):E2314-2319.
[19] Takatani R,Minagawa M,Molinaro A,et al. Similar frequency of paternal uniparental disomy involving chromosome 20q (patUPD20q) in Japanese and Caucasian patients affected by sporadic pseudohypoparathyroidism type Ib(sporPHP1B)[J].Bone,2015,79:15-21.[20] Dixit A,Chandler KE,Lever M,et al.Pseudohypoparathyroidism type Ib due to paternal uniparental disomy of chromosome 20q[J].J Clin Endocrinol Metab,2013,98(1):E103-108.
[21] Elli FM,de Sanctis L,Bollati V,et al.Quantitative analysis of methylation defects and correlation with clinical characteristics in patients with pseudohypoparathyroidism type I and GNAS epigenetic alterations[J].J Clin Endocrinol Metab,2014,99(3):E508-517.
[22] Brix B,Werrner R,Staedt P,et al.Different pattern of epigenetic changes of the GNAS gene locusin patenets with pseudohypoparathyroidism type Ic confirm the heterogeneity of underlying pathomechanisms in this subgroup of pseudohypo-parathyroidism and the demand for a new classification of GNAS-related disorders[J].J Clin Endocrinol Metab,2014,99(8):E1564-1570.
Pseudohypoparathyroidism and hypokalemia: clinical manifestation analysis
Zhang Wei1,2, Zhang Mei1, Wang Yucheng1, Cui Dai1, Zhou Hongwen1, Chen Huanhuan1, Yuan Qingxin1, Li Jianbo1, Yang Tao1
1.DepartmentofEndocrinologyandMetabolism,theFirstAffiliatedHospitalofNanjingMedicalUniversity,Nanjing210029,China;2.DepartmentofEndocrinologyandMetabolism,theSceondPeople'sHospitalofWuhu,Wuhu241000,China
ZhangMei,Email:zhangmei@njmu.edu.cn
Objective To investigate the clinical characteristics of patients with pseudohypoparathyroidism(PHP) and hypokalemia. Methods The clinical data of 13 patients with PHP in our hospital from July 2006 to April 2016 were analyzed retrospectively. Results None of which had Albright's hereditary osteodystrophy(AHD). Convulsion was observed in 86.4% patients. Myasthenia and numbness of limbs were 38.5%. Hypocalcemia, hyperphospheremia and high level of parathyroid hormone was the main characteristics of laboratory examination. In all 13 cases, hypokalemia rate was 46.2%,10 with abnormal thyroid function. The course of the disease was longer in patients with hypokalemia than controls. There was no significant difference in other clinical manifestation and laboratory examination in patients with or without hypokalemia. There were no typical AHO body deformity. The clinical manifestations of 84.6% patients were tetany symptoms and 38.5% patients had limb weakness and numbness. 6 patients were with hypokalemia. 10 patients with abnormal thyroid function. The patients with hypokalemia had longer course of disease. The serum potassium was between 3-3.5 mmol/L and other clinical and biochemical indicators and non hypokalemia group had no significant difference. Conclusion PHP patients were susceptible to hypokalemia and subclinical hypothyroidism.
pseudohypoparathyroidism;hypokalemia; hypothyroidism
江苏省临床医学科技专项(BL2012026)
张梅, Email: zhangmei@njmu.edu.cn
R582.2
A
1004-583X(2017)09-0759-04
10.3969/j.issn.1004-583X.2017.09.006
2017-06-26 编辑:张卫国
