戈谢病GBA基因突变致血小板输注无效1例*
2017-06-19吴红江梅闻芳聂益军
吴红,江梅,闻芳,聂益军
(1.江西省血液中心输血研究室,南昌 330052;2.南昌大学第一附属医院检验科,南昌 330006)
·临床实验研究·
戈谢病GBA基因突变致血小板输注无效1例*
吴红1,江梅2,闻芳2,聂益军2
(1.江西省血液中心输血研究室,南昌 330052;2.南昌大学第一附属医院检验科,南昌 330006)
目的 分析1例血小板输注无效患者及其家系戈谢病葡萄糖脑苷脂酶(GBA)基因突变特征。方法 1例贫血及血小板减少的女性患者经血小板输注无效后,对其进行骨髓细胞学、B超及基因测序检测;采用干血斑法检测其白细胞β-葡糖脑苷脂酶活性;提取该家系8个成员(包括先证者及其直系亲属)外周血基因组DNA进行基因测序。结果 骨髓细胞学检查示该例患者可见戈谢细胞(6.0%),白细胞β-葡糖脑苷脂酶活性为3.78 nmol/(h·mgPro),B超结果示脾肿大;基因测序分析发现其为GBA基因c.484A>G纯合错义突变;家系调查结果表明,先证者父母、子女及妹妹5人均为GBA基因杂合突变。结论 戈谢病患者可因脾功能亢进而出现血小板输注无效;GBA基因突变为该家系的主要致病因素。
血小板输注无效;戈谢病;葡萄糖苷酯酶基因突变
戈谢病(Gaucher disease gaucherdisease)即葡糖脑苷脂酶病,是一种家族性糖脂代谢疾病,为常染色体隐性遗传,其主要临床表现为肝脾肿大、贫血及血小板减少[1]。目前国内外对戈谢病分子机制的研究多集中在葡萄糖脑苷脂酶(GBA)基因突变上[2-3]。GBA基因突变的类型可因种族或地区的不同而呈现出不同的特点[4]。本研究通过对1例血小板输注无效患者及其家系进行调查,探讨其GBA基因突变的特征。
1 材料与方法
1.1 临床资料及家系调查 先证者,女性,38岁,有2~3年的贫血史,其他病史不详。2016年11月在某县级医院检查PLT为47×109/L,输注机采血小板制品2 U,输注后24 h其血小板回收率<16%(即输注无效)。2016年12月1日遂前往南昌大学第一附属医院做进一步检查(血液检查、骨髓细胞学、X光胸透及B超)。其影像学结果示:X光胸透检查正常;B超显示肝回声均匀、致密;脾肿大(6.2 cm×25 cm)。且该患者呈进行性脾肿大(3个月后脾大小为7.1 cm×26 cm)。由于经费等原因,先证者接受部分医学检测(如基因测序),但拒绝进行同位素骨扫描及医学治疗。询问先证者的疾病家族史,并对该家系8名(包括先证者、其父母亲、其弟及其妹、其配偶、其子及其女)人员进行临床资料的收集。本研究经医院伦理学委员会批准,患者及家系成员知情同意。
1.2 主要仪器与试剂 全自动血细胞分析仪及配套试剂(Sysmex XE2100,日本Sysmex公司);全自动
生化仪(AU5400,美国贝克曼库尔特公司);酶联仪(PHOMO,郑州安图生物公司):全自动核酸提取仪(MagCoreHF16,上海吉盈公司);热循环仪PCR仪(美国ABI公司);Agilent 2100 生物分析仪(美国安捷伦公司);BES4000测序仪(北京博奥公司);生化检测试剂盒(上海科华公司);DNA提取试剂盒(RBCBioscience,上海吉盈公司)。
1.3 标本采集及实验室检测 采集该例患者入院时的空腹静脉血标本2 mL,EDTA-K2抗凝,用全自动血细胞分析仪及配套试剂检测该患者血液学指标。另取非抗凝全血5 mL,室温静置1~2 h,2 500 r/min离心10 min,分离血清,用全自动生化仪及相应的生化检测试剂盒检测总胆红素(T-Bil)、直接胆红素(D-Bil)、血清铁(serum iron)、总蛋白(TP)、清蛋白(Alb)、天门冬氨酸氨基转移酶(AST)、丙氨酸氨基转移酶(ALT)、维生素B12、叶酸等指标。其余指标均由南昌大学第一附属医院检验科检测。由临床医师采集骨髓标本后,行瑞氏-吉姆萨染色,光学显微镜下观察骨髓细胞形态学并拍照。
1.4 干血斑法检测白细胞β-葡糖脑苷脂酶活性 取患者新鲜全血(约50 μL)滴在滤纸上,获得直径约1 cm 血斑,于室温静置4 h直至干燥,置于密封塑料袋中送至广州市妇女儿童医疗中心检测白细胞β-葡糖脑苷脂酶活性并进行结果判读。
1.5 基因测序
1.5.1 DNA提取 采集该家系8名(包括先证者、其父母亲、其弟及其妹、其配偶、其子及其女)人员空腹静脉血2 mL,枸橼酸钠抗凝,置-80 ℃保存。采用MagCoreHF16全自动核酸提取仪及DNA提取试剂盒提取DNA,样本置-20 ℃保存。
1.5.2 探针设计及高通量基因测序 采用SureDesign在线设计工具(美国安捷伦公司)进行探针设计使,捕获探针由美国安捷伦公司采用SureDesign在线设计工具设计并合成。取提取的DNA样本,送至东莞博奥木华公司,用Ion PITMChip v3和BSE4000测序仪进行高通量测序。采用TMAP(Torrent Mapping Alignment Program)将测序序列比对到人类参考基因组(HG19, NCBI release GRCh37),并利用TVC(Torrent Variant Caller)分析突变,使用ANNOVAR软件对突变进行注释,包括注释突变的位置、突变的基因信息、质量得分、氨基酸改变、SIFT和PolyPhen-2功能性预测、dbSNP数据库(https://www.ncbi.nlm.nih.gov/snp/)和ExAC数据库(http://exac.broadinstitute.org/)人群频率等。共检测17种遗传病(包括进行性假肥大性肌营养不良、甲型及乙型血友病、α,β地中海贫血、遗传性耳聋、脊髓性肌肉萎缩症、半乳糖血症、苯丙酮尿症、肝豆状核变性、先天性肾上腺皮质增生症、糖原累积病Ⅱ型、瓜氨酸血症、戈谢病、白化病、粘多糖I型及Ⅱ型、多囊肾),涉及26个基因(包括DMD、F8、F9、HBA1、HBA2、HBB、GJB2、SLC26A4、GJB3、SMN1、GALT、PAH、ATP7B、CYP21A2、GAA、ASS1、SLC25A13、GBA、GPR143、TYR、TYRP1、OCA2、SLC45A2、IDUA、IDS、PKHD1)。
1.5.3 第一代基因测序 对GBA基因的PCR扩增产物进行Sanger测序(由Thermo Fisher公司完成),并将测序结果与GenBank文库中的参考序列进行BLAST比对分析。
2 结果
2.1 先证者家系遗传特征 该家系3代共8名,其中男性4名,女性4名。该先证者为GBA基因的纯合突变,其临床表现为贫血及血小板减少。先证者的直系亲属均无明显的临床症状(如贫血及血小板减少);先证者的父亲和母亲为亲表兄妹关系(即近亲结婚)。对先证者的家人进行测序,结果发现:先证者的父亲、母亲、儿子、女儿、妹妹均为GBA基因的杂合突变;其丈夫及弟弟则为健康者。家系结果见图1。
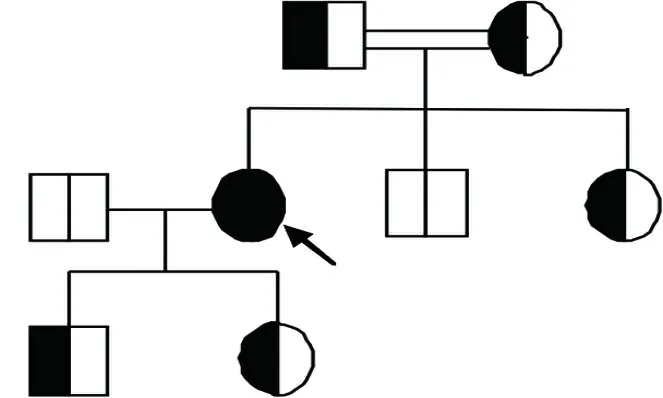
注:,常染色体隐性基因男性携带者;,正常男性;,常染色体隐性基因女性携带者;,女性先证者;,近亲结婚。
图1 该先证者家系图谱
2.2 血液学检测结果 该例患者的红细胞、血红蛋白、血小板及白细胞β-葡糖脑苷脂酶活性均低于参考范围;而总胆红素、直接胆红素、血沉、血清铁蛋白、IgG、IgM及抗-DNA抗体均高于参考范围。见表1。
2.3 骨髓细胞学结果 骨髓涂片(瑞氏-吉姆萨染色)示:粒、红系增生活跃,粒、红比为1.7∶1;戈榭细胞约占6.0%,此类细胞体积较大,形态不规则,多呈圆形、卵圆形,含有1个或数个偏位的细胞核,核染色质粗糙,胞浆量多,呈淡蓝色,浆内充满交织成网状或洋葱皮样的条纹结构。见图2。
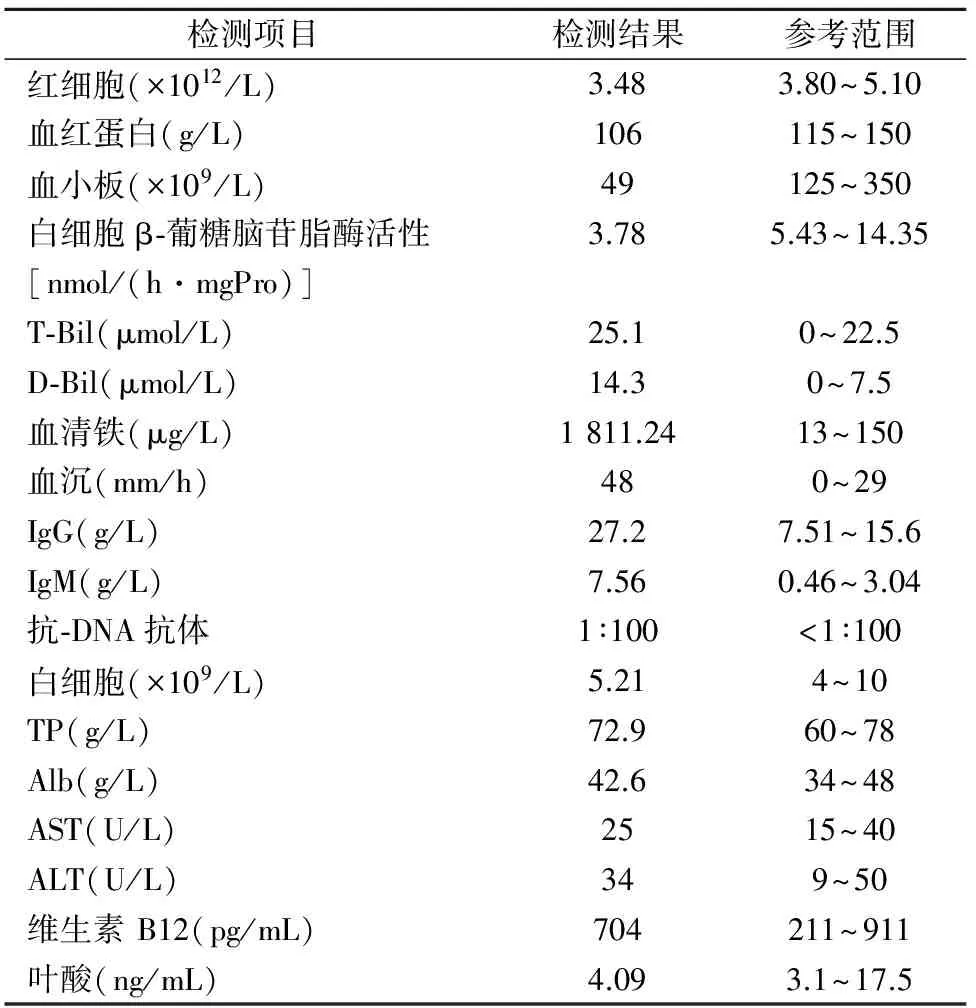
表1 该例患者血液学检测结果
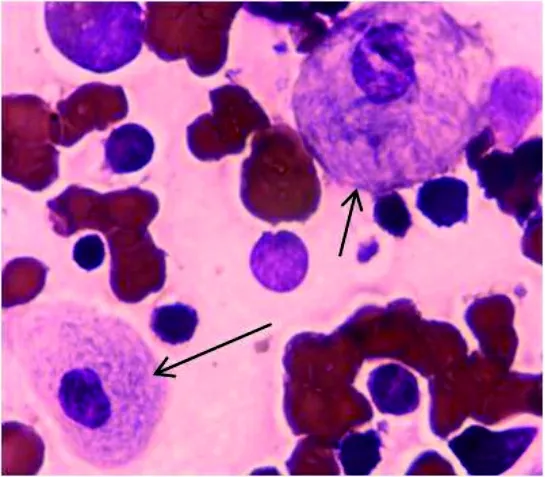
注:箭头为戈谢细胞(瑞氏-吉姆萨染色)。
图2 骨髓细胞学检测结果(×1 000)
2.4 基因测序结果 先证者携带1个临床意义未明突变:GBA基因c.484A>G 纯合错义突变,家系分析发现其父亲、母亲、儿子、女儿及妹妹均为GBA基因c.484A>G 杂合错义突变,而其丈夫、弟弟则为健康人群(GBA基因野生型)。测序结果见图3。
3 讨论
戈谢病是一种溶酶体贮积病(常染色体隐性遗传病),由于戈谢细胞在脾脏中积累引起脾肿大,造成脾功能亢进,导致血小板输注无效[5-7]。本研究中的患者出现脾肿大,且红细胞及血小板同时减少、骨髓细胞增生活跃,因此该患者为脾功能亢进症,致血小板输注无效。基于该患者骨髓中检出戈谢细胞(图2)、白细胞β-葡糖脑苷脂酶活性降低(表1)及其临床症状,综合诊断该成年女性患者为戈谢病Ⅰ型。研究表明,约70%以上的戈谢病Ⅰ型患者存在骨骼损害及生长迟缓等症状[8],但该患者拒绝接受骨密度或骨扫描的检查,因此未能确定其是否存在骨骼病变。
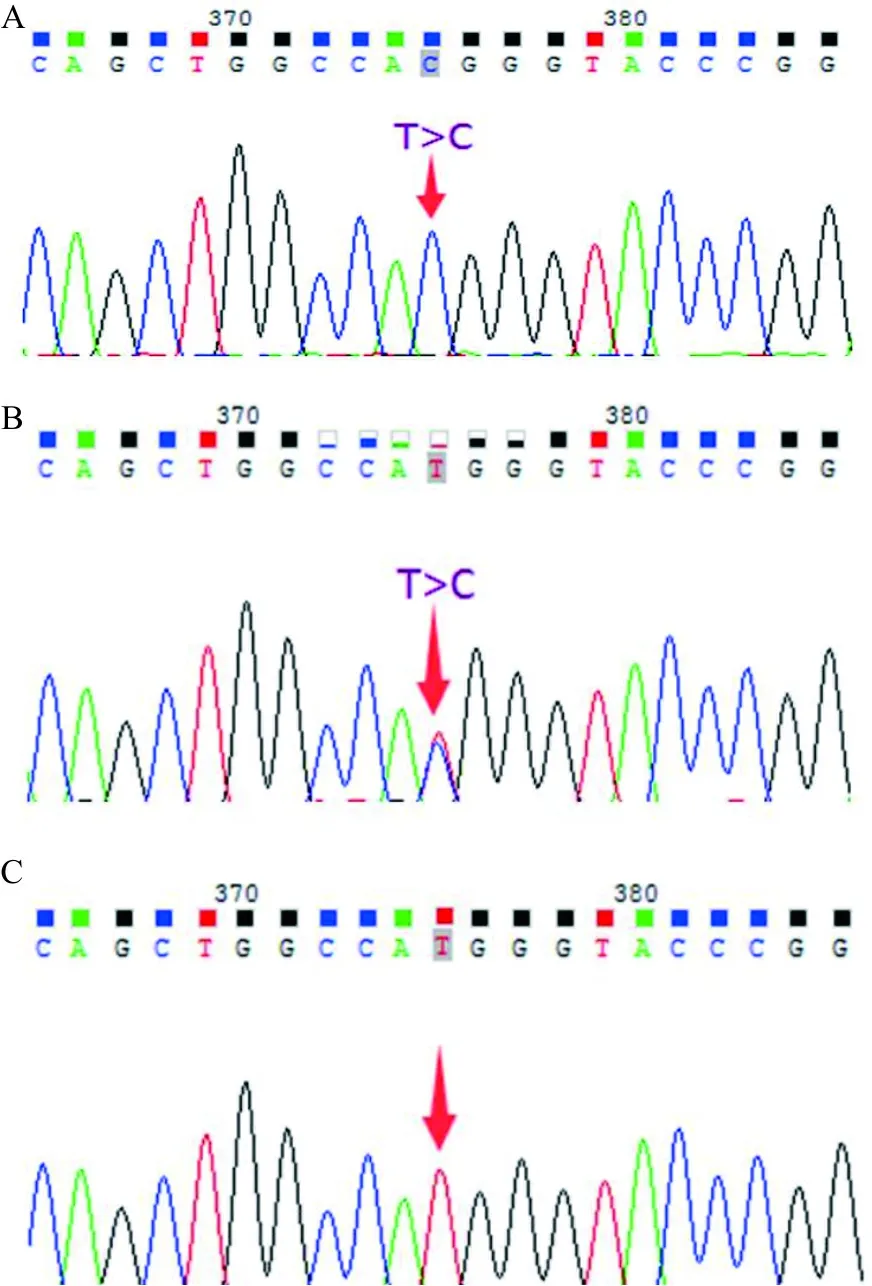
注:A,GBA基因:c.484A>G 纯合错义突变;B,GBA基因: c.484A>G 杂合错义突变;C,GBA基因野生型;箭头示突变位点。
图3 该家系GBA基因测序结果
GBA是一种可溶性的糖脂类物质,GBA基因突变造成单核-巨噬细胞内的葡萄糖脑苷脂不能被水解从而形成典型的戈谢细胞[9]。目前已发现超过350种GBA基因突变类型(包括错义突变,无义突变,移码突变的插入或缺失突变,剪接突变及复杂等位基因突变),其中2种GBA基因错义突变类型p.L444P(c.1448T>C)和 p.N370S(c.1226A>G)是戈谢病患者最常见的基因突变类型[10-11]。GBA基因突变可在不同的种族人群中出现不同的特征,如在犹太人群中以4种GBA基因突变类型(N370S,L444P,84insG和IVS2+1G>A)为主(占90%),而在高加索人群中以6种GBA基因突变类型(N370S,L444P,RecNciI,RecTL,D409H和IVS2+1G>A)为主(占60%~70%)[10-11]。本研究中发现的GBA突变: c.484A>G 纯合错义突变可导致其编码的肽链第 162 号甲硫氨酸的密码子变成缬氨酸(p.M162V),该突变人群频率极低。
为进一步了解该GBA基因突变的产生原因,本研究对先证者进行GBA基因家系调查。结果表明,先证者的父亲、母亲、儿子、女儿、妹妹均为GBA基因的杂合突变;其丈夫及弟弟则为健康者。因为先证者的父亲和母亲为亲表兄妹关系,所以推测该GBA基因突变可能源自其父母的某位共同祖先。根据该先证者的临床表现,可推定该GBA基因突变(c.484A> G)为轻度和非神经元病变的性质,与戈谢病Ⅰ型相关,此纯合突变在亚洲人群中极为罕见。本次对戈谢病Ⅰ型及GBA基因突变的研究尚存在不足之处,主要包括(1)未能对该患者进行更多的医学检测(如骨密度或骨扫描);(2)由于该患者拒绝临床治疗,未能观察其治疗后的相关指针;(3)未能对GBA突变基因在细胞水平下进一步验证。
[1]考杉斯基.威廉姆斯血液学[M].第8版.陈竺等译.北京:人民卫生出版社, 2011: 988-994.
[2]Amico G, Grossi S, Vijzelaar R,etal. MLPA-based approach for initial and simultaneous detection of GBA deletions and recombinant alleles in patients affected by Gaucher Disease[J]. Mol Gene Metab, 2016, 119(4):329-337.
[3]陈健, 孟岩, 石秀玉,等. 戈谢病Ⅲ型同胞患儿临床及GBA基因突变分析[J]. 临床儿科杂志, 2015, 33(5):462-465.
[4]Stirnemann J, Belmatoug N, Camou F,etal. A review of gaucherdisease pathophysiology, clinical presentation and treatments[J]. Int J Mol Sci, 2017, 18(2):441-470.
[5]胡蜀红, 陈琛, 朱文君,等. 成年女性Ⅰ型戈谢病1例并文献复习[J]. 内科急危重症杂志, 2015, 21(6):470-472.
[6]Rosenbloom BE, Weinreb NJ. Gaucher disease: a comprehensive review[J]. Crit Rev Oncogenesis, 2013, 18(3):163-175.
[7]崔婷婷, 孙志霞, 李殿秋,等. 脾戈谢病超声表现1例[J]. 中国医学影像技术, 2016, 32(4):508.
[8]Mistry PK, Belmatoug N, Vom Dahl S,etal. Understanding the natural history of Gaucher disease[J]. Am J Hemato, 2015, 90 (S1):S6-S11.
[9]刘林玉, 杜司晨, 张进,等. 戈谢氏病致病机制及治疗方法[J]. 遗传, 2015, 37(6):510-516.
[10]Hoitsema K, Amato D, Khan A,etal. Identification of novel splice site mutation IVS9+1(G > A) and novel complex allele G355R/R359X in Type 1 Gaucher patients heterozygous for mutation N370S[J]. Meta Gene, 2016, 9(C):47-51.
[11]Mattošová S, Chandoga J, Hlavatá A,etal. Spectrum ofGBAmutations in patients with Gaucher disease from Slovakia: identification of five novel mutations[J]. IMAJ, 2015, 17(3):166-170.
(本文编辑:许晓蒙)
Platelet transfusion refractoriness caused byGBAgene mutation in one patient with Gaucher disease
WUHong1,JIANGMei2,WENFang2,NIEYi-jun2
(1.DepartmentofBloodTransfusion,JiangxiProvinceBloodCenter,Nanchang330052,Jiangxi; 2.DepartmentofClinicalLaboratory,theFirstAffliatedHospitalofNanchangUniversity,Nanchang330006,Jiangxi,China)
Objective To analyze the mutation characteristics ofGBAgene in one patient with Gaucher disease and platelet transfusion refractoriness. Methods A female patient with anemia and thrombocytopenia showed platelet transfusion refractoriness, and then the proband and her family were performed bone marrow smear, β-glucocerebrosidase activity in leukocytes (dried blood spot assay), B-ultrasonography and gene sequencing examination and pedigree investigation. Results Pedigree investigation showed that the heterozygous mutation ofGBAgene existed in the father, mother, son, daughter and sister of the proband. Bone marrow cytomorphologic examination showed that Gaucher cells accounted for 6.0% in the female patient. The β-glucocerebrosidase activity in leukocytes was 3.78 nmol/(h·mg Pro). B-ultrasonography showed slightly splenomegaly. Gene sequencing found that the homozygous mutation ofGBAgene, c.484A>G, existed in the female patient. Conclusion The patients with Gaucher disease may appear platelet transfusion refractoriness due to hypersplenism. The mutation ofGBAgene is the main pathogenic factor of the family with Gaucher disease.
platelet transfusion refractoriness; Gaucher disease;GBAgene mutation
10.13602/j.cnki.jcls.2017.05.03
国家自然科学基金(81660144)。
吴红,1975年生,女,副主任技师,博士,主要从事输血相关技术。
聂益军,副主任技师,E-mail:wuhongnie@163.com。
R331
A
2017-03-01)
