Gerstmann-Sträussler-Scheinker病1例报告
2016-12-22赵明明申平平冯加纯
赵明明, 王 爽, 侯 帅, 申平平, 冯加纯
Gerstmann-Sträussler-Scheinker病1例报告
赵明明, 王 爽, 侯 帅, 申平平, 冯加纯
朊蛋白病,又称感染性海绵状脑病,是由一种缺少核酸但具有感染性的朊蛋白引起的致死性的神经变性疾病,分为散发性、遗传性及获得性。在人类,遗传性朊蛋白病占朊蛋白病的5%~15%,包括家族性Creutzfeldt-Jakob 病(Creutzfeldt-Jakob disease,CJD)、Gerstmann-Sträussler-Scheinker(GSS)病以及家族性致死性失眠症(fatal familial insomnia,FFI)[1]。其中,GSS病(人类孟德尔遗传 137440)是常染色体显性遗传,发病率仅有每年1~10/108[2]。临床上,该病早期主要以渐进性小脑共济失调为主,伴有锥体束征及锥体外系损伤表现,晚期可出现严重痴呆[3~8]。在我国已报道的确诊的GSS病仅有4个家系[6,9~11]。现将1例经基因诊断的GSS病报道如下。
1 病例资料
患者,男,50岁,因走路不稳2.5 y,加重伴言语笨拙、饮水呛咳1 y于2015年2月入院。患者于2.5 y前无明显诱因逐渐出现走路不稳、双足拖地,自觉双下肢僵硬,不伴疼痛或麻木。入院前2 y出现下肢冰凉感由双足蔓延至大腿根部。入院前1.5y出现全身乏力,写字异常,表现为字越写越小。入院前1 y出现言语欠流利及饮水呛咳,于外院行SCAs基因检测未见异常,诊断为“脊髓小脑性共济失调可能性大”,给予对症治疗。上述症状逐渐加重,并逐渐出现头部不自主震颤,坐位不稳,自觉双下肢肌肉酸痛难忍,遂就诊于我院。病程中无尿便障碍,体重未见明显变化。病前无感染及外伤史,无特殊毒物及药物接触史,无牛羊接触史。既往体健。家族史:患者的母亲及长兄均有相似的症状,已故(见图1、表1)。
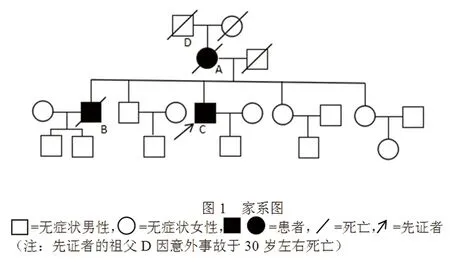
图1 家系图

患者性别发病年龄(岁)病程首发症状主要临床表现辅助检查病理基因A(母亲)B(长兄)C(先证者)女男男5136475y9y>4y共济失调共济失调共济失调构音障碍,吞咽困难,双下肢肌肉萎缩,晚期严重痴呆构音障碍,吞咽困难,肌肉萎缩(下肢为著),震颤晚期轻微痴呆及尿便障碍构音障碍,饮水呛咳,双下肢肌肉萎缩及疼痛,双下肢腱反射消失,双侧病理征阳性,偶尿失禁,震颤,尚未出现认知障碍头部CT:小脑萎缩不详详见病例资料未检未检未检未检未检SCAs基因阴性PRNP基因P102L突变
查体:血压122/76 mmHg,心率76次/min。神清,构音障碍。记忆力、定向力、计算力正常,双侧瞳孔等大同圆,直径约3.0 mm,直间接对光反射灵敏,双侧眼球向各方向运动灵活,无眼震。双上肢肌力5级,双下肢肌力5-级,四肢肌张力正常,双上肢腱反射对称引出,双下肢腱反射未引出,深浅感觉未见异常,指鼻试验欠稳准,双手轮替动作笨拙,双侧跟膝胫试验欠稳准,睁闭眼时均站立不稳,宽基步态。双侧Babinski及Chaddock征阳性。入院前2.5 y行腰椎CT未见明显异常。入院前2 y行头部、颈椎、胸椎、腰椎MRI及肌电图未见异常。入院前1 y于外院辅助检查,血常规、尿常规、便常规、凝血常规、生化、蛋白电泳、甲状腺功能、血沉、类风湿因子、抗O抗体、肿瘤标志物、甲状腺超声、双下肢静脉超声、头MRI、肌电图、平衡功能检查均未见异常;血脂:总胆固醇5.93 mmol/L,低密度脂蛋白胆固醇 4.3 mmol/L;汉密顿焦虑量表:11分,轻度焦虑状态;汉密顿抑郁量表:15分;黑质超声:强度II级,中脑面积约4.95 cm;腰椎MRI:L4-5、L5-S1腰椎间盘膨出,腰椎退行性病变;SCAs基因检测未见异常。入院后辅助检查,低密度脂蛋白胆固醇3.64 mmol/L;脑脊液免疫球蛋白IgG 40.60 mg/l,脑脊液蛋白0.61 g/L,潘氏反应+,NMO-IgG(血、脑脊液)、AQP-4-Ab(血、脑脊液)、副肿瘤抗体(血、脑脊液)、MBP(脑脊液):阴性(-);肿瘤标志物未见异常;头部MRI:脑内MR平扫未见明显异常信号;肌电图:双下肢所测神经肌肉均未见明显异常。采静脉血行基因测序,于PRNP基因外显子序列发现一处杂合突变位点:c. 305C>T(胞嘧啶>胸腺嘧啶),导致氨基酸改变:p. P102L(脯氨酸>亮氨酸),考虑GSS病诊断明确。出院后随访1.5 y,现患者共济失调、构音障碍、饮水呛咳进行性加重,出现明显震颤及双下肢肌肉萎缩、疼痛,便秘,偶有尿失禁,日常生活无法自理。建议患者复查头MRI、脑电图、肌电图及腰椎穿刺术,建议直系亲属进行PRNP基因测序,患者及家属表示暂拒绝。
2 讨 论
Gerstmann-Sträussler-Scheinker(GSS)病是由位于20号染色体的朊蛋白基因(PRNP)发生突变[12],使其编码的正常存在于健康人体中枢神经系统细胞膜上的朊蛋白(PrPc)转变为不溶性的朊蛋白(PrPsc)沉积在胞内而导致的遗传性中枢神经系统变性疾病[13]。1936年神经病理学家Gerstmann、Sträussler及Scheinker首次报道了奥地利的一个家系并命名为GSS病[14],1991年Kretzschmar等[15]在该家系中检测到PRNP基因P102L突变。我国首例GSS病于1993年由林世和教授经脑组织活检病理诊断[11]。GSS病通常在50岁左右发病,是病程最长的一类朊蛋白病,平均约5~7 y,最长达12 y[2]。典型的临床表现为进行性小脑共济失调,伴锥体束征、假性球麻痹、肌阵挛及腱反射消失,多数情况下晚期可有痴呆[3~8]。脑组织病理是诊断该病的重要依据,主要表现为大脑皮质、小脑皮质及基底节局部多中心淀粉样蛋白斑块沉积,神经元缺失及星形胶质细胞增生,伴或不伴海绵状变化、神经元纤维缠结及tau蛋白沉积[1,3,5,16]。病变组织行Western blot可检测到6Kd到10Kd蛋白条带,为同时在C-末端及N-末端截短的非糖基化的具有蛋白酶抗性的PrPsc片段[16~18]。
近年来,基因检测逐步成了诊断GSS病的最重要手段,迄今为止已有超过30个PRNP突变位点(包括错义突变及无义突变)在多个国家被报道[4,12,19~21],如P102L、P105L、F198S、A117V、Q217R等,其中P102 L(脯氨酸被亮氨酸取代)是最常见的突变[19,22]。PRNP的基因型与临床表型具有高度的异质性[23~26],即使具有相同突变类型,其临床表型也差异显著,如Hsiao等[25]报道A117V的患者表现为老年痴呆及锥体外系症状,而Mastrianni等[27]报道的家族表现为共济失调而非痴呆。就P102L来说,已报道的临床表型有:典型的小脑性共济失调、精神症状[28]、周围神经改变[6]、迅速进展的痴呆和缓慢渐进性共济失调[29]等类型。其分子机制可能部分取决于129密码子甲硫氨酸/缬氨酸(M/V)上等位基因的多态性,而这尚存争议[29~31]。
目前,本病诊断主要依据临床表现、阳性家族史、脑组织病理及基因检测。本例患者以共济失调起病,有锥体束征及锥体外系表现,并逐渐出现假性球麻痹,有阳性家族史,结合基因检测结果,可明确诊断GSS病。本文报道的病例有明确的双下肢腱反射消失与我国台湾及日本十余个家系表型一致[10,32],而我国神经病理学家林世和教授等[11]于1993年经病理学确诊的GSS病患者及Takanori等[33]于2010年报道的一例日本典型P102L突变患者则表现为腱反射亢进,Yamada等[34]认为朊蛋白沉积在腰髓后角导致了腱反射消失,而腱反射亢进则是由于朊蛋白沉积在脊髓后角之前大脑皮质及皮质脊髓束的损伤导致[33]。据文献报道,头部MRI在病程早期通常是正常的,随病程进展可出现大脑和(或)小脑半球萎缩,这种改变可能和朊蛋白与铁、铜、锰的结合有一定关系[35]。本例患者发病2.5 y内3次行头MRI及肌电图均未见异常可能与病程早期有关。有研究认为质子磁共振波谱分析(1H-MRS)可能对早期GSS病的诊断具有提示作用[36]。
本例患者有典型的小脑共济失调症状且尚未出现痴呆,极易被误诊为脊髓小脑性共济失调。本例患者SCA全套基因筛查均为阴性,扩大基因筛查范围使得最终确诊为GSS病。同样,在意大利206例SCA及Friedreich共济失调基因均为阴性的共济失调患者中查到7例PRNP基因突变[37]。这提示我们对于SCA检测阴性的共济失调患者应该考虑此病。由于高度的临床异质性使GSS病的诊断具有挑战性,需与多种疾病相鉴别。John等[38]报道一例临床表现为非典型额颞叶痴呆的患者,在发病8 y及10 y先后被诊断为Pick病、皮质基底节变性,发病13 y死亡后经病理及基因检测明确诊断为GSS病Q217R型。Yuval等[39]报道的一例具有典型共济失调及构音障碍经辅助检查诊断为多发性硬化的患者,多种治疗无效,症状持续进展,1.5 y后结合其阳性家族史并经基因检测最终确诊为GSS病。Webb等[31]对英国近百例P102L患者的研究表明,一部分以认知障碍及精神症状为特征且脑脊液14-3-3蛋白及脑电图均异常的患者,极易被误诊为散发型CJD,而少部分以痴呆起病、头MRI表现为T2皮质下深部白质高信号的患者极易被误诊为Binswanger病。由此,基因检测为该病的诊断提供了极其重要的依据。
3 总 结
由于GSS病在我国极为罕见,具有典型GSS病临床表现的患者应考虑到此病的可能性。此外,导致GSS病的PRNP基因突变位点众多,临床表型多样,单纯依靠典型的临床表现及阳性家族史诊断该病仍有局限性,基因检测对GSS病的确诊具有重要作用。该病基因型-表型的关系及机制尚不确切,需进一步深入研究。该病死亡率100%,目前尚无有效的治疗方法,基因治疗是今后的研究方向。临床工作中,应注意加强身心护理、避免感染,给予对症治疗,提高患者生存质量。
[1]Gambetti P,Kong Q,Zou W,et al. Sporadic and familial CJD:classification and characterisation[J]. British Medical Bulletin,2003,66:213-239.
[2]Kovacs GG,Puopolo M,Ladogana A,et al. Genetic prion disease:the EUROCJD experience[J]. Human Genetics,2005,118(2):166-174.
[3]Masters CL,Gajdusek DC,Gibbs CJ. Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Straussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus-induced spongiform encephalopathies[J]. Brain:A Journal of Neurology,1981,104(3):559-588.
[4]Collins S,McLean CA,Masters CL. Gerstmann-Straussler-Scheinker syndrome,fatal familial insomnia,and kuru:a review of these less common human transmissible spongiform encephalopathies[J]. Journal of Clinical Neuroscience:Official Journal of the Neurosurgical Society of Australasia,2001,8(5):387-397.
[5]Piccardo P,Dlouhy SR,Lievens PM,et al. Phenotypic variability of Gerstmann-Straussler-Scheinker disease is associated with prion protein heterogeneity[J]. Journal of Neuropathology and Experimental Neurology,1998,57(10):979-988.
[6]Young K,Jones CK,Piccardo P,et al. Gerstmann-Straussler-Scheinker disease with mutation at codon 102 and methionine at codon 129 of PRNP in previously unreported patients[J]. Neurology,1995,45(6):1127-1134.
[7]Ghetti B,Dlouhy SR,Giaccone G,et al. Gerstmann-Straussler-Scheinker disease and the Indiana kindred[J]. Brain Pathology,1995,5(1):61-75.
[8]Gambetti P,Parchi P,Chen SG. Hereditary Creutzfeldt-Jakob disease and fatal familial insomnia[J]. Clinics in Laboratory Medicine,2003,23(1):43-64.
[9]Ye J,Han J,Shi Q,et al. Human prion disease with a G114V mutation and epidemiological studies in a Chinese family:a case series[J]. Journal of Medical Case Reports,2008,2:331.
[10]Chi NF,Lee YC,Lu YC,et al. Transmissible spongiform encephalopathies with P102L mutation of PRNP manifesting different phenotypes:clinical,neuroimaging,and electrophysiological studies in Chinese kindred in Taiwan[J]. Journal of Neurology,2010,257(2):191-197.
[11]林世和,江新梅,赵节绪,等. Gerstmann-Strāussler综合征-一个家系两代5例临床、病理、免疫组化及动物接种传递的实验研究[J]. 临床神经病学杂志,1993,4:200-202.
[12]Doh-ura K,Tateishi J,Sasaki H,et al. Pro-leu change at position 102 of prion protein is the most common but not the sole mutation related to Gerstmann-Straussler syndrome[J]. Biochemical and Biophysical Research Communications,1989,163(2):974-979.
[13]Ironside JW,Head MW. Biology and neuropathology of prion diseases[J]. Hand Clin Neurol,2008,89:779-797.
[14]Hainfellner JA,Brantner-Inthaler S,Cervenakova L,et al. The original Gerstmann-Straussler-Scheinker family of Austria:divergent clinicopathological phenotypes but constant PrP genotype[J]. Brain Pathology,1995,5(3):201-211.
[15]Kretzschmar HA,Honold G,Seitelberger F,et al. Prion protein mutation in family first reported by Gerstmann,Straussler,and Scheinker[J]. Lancet (London,England),1991,337(8750):1160.
[16]Ghetti B,Tagliavini F,Takao M,et al. Hereditary prion protein amyloidoses[J]. Clinics in Laboratory Medicine,2003,23(1):65-85.
[17]Parchi P,Chen SG,Brown P,et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Straussler-Scheinker disease[J]. Proceedings of the National Academy of Sciences of the United States of America,1998,95(14):8322-8827.
[18]Tagliavini F,Prelli F,Ghiso J,et al. Amyloid protein of Gerstmann-Straussler-Scheinker disease (Indiana kindred) is an 11 kd fragment of prion protein with an N-terminal glycine at codon 58[J]. The EMBO Journal,1991,10(3):513-519.
[19]Hsiao K,Baker HF,Crow TJ,et al. Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome[J]. Nature,1989,338(6213):342-345.
[20]Hill AF,Joiner S,Beck JA,et al. Distinct glycoform ratios of protease resistant prion protein associated with PRNP point mutations[J]. Brain:A Journal of Neurology,2006,129(Pt 3):676-685.
[21]Parchi P,Saverioni D. Molecular pathology,classification,and diagnosis of sporadic human prion disease variants[J]. Folia Neuropathologica / Association of Polish Neuropathologists and Medical Research Centre,Polish Academy of Sciences,2012,50(1):20-45.
[22]Liberski PP. Gerstmann-Straussler-Scheinker disease[J]. Advances in Experimental Medicine and Biology,2012,724:128-137.
[23]Kitamoto T,Iizuka R,Tateishi J. An amber mutation of prion protein in Gerstmann-Straussler syndrome with mutant PrP plaques[J]. Biochemical and Biophysical Research Communications,1993,192(2):525-531.
[24]Ghetti B,Tagliavini F,Masters CL,et al. Gerstmann-Straussler-Scheinker disease. II. Neurofibrillary tangles and plaques with PrP-amyloid coexist in an affected family[J]. Neurology,1989,39(11):1453-14561.
[25]Hsiao KK,Cass C,Schellenberg GD,et al. A prion protein variant in a family with the telencephalic form of Gerstmann-Straussler-Scheinker syndrome[J]. Neurology,1991,41(5):681-684.
[26]Synofzik M,Bauer P,Schols L. Prion mutation D178N with highly variable disease onset and phenotype[J]. Journal of Neurology,Neurosurgery,and Psychiatry,2009,80(3):345-346.
[27]Mastrianni JA,Curtis MT,Oberholtzer JC,et al. Prion disease (PrP-A117V) presenting with ataxia instead of dementia[J]. Neurology,1995,45(11):2042-2050.
[28]Majtenyi C,Brown P,Cervenakova L,et al. A three-sister sibship of Gerstmann-Straussler-Scheinker disease with a CJD phenotype[J]. Neurology,2000,54(11):2133-2137.
[29]Barbanti P,Fabbrini G,Salvatore M,et al. Polymorphism at codon 129 or codon 219 of PRNP and clinical heterogeneity in a previously unreported family with Gerstmann-Straussler-Scheinker disease (PrP-P102L mutation)[J]. Neurology,1996,47(3):734-741.
[30]Giovagnoli AR,Di Fede G,Aresi A,et al. Atypical frontotemporal dementia as a new clinical phenotype of Gerstmann-Straussler-Scheinker disease with the PrP-P102L mutation. Description of a previously unreported Italian family[J]. Neurological Sciences:Official Journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology,2008,29(6):405-410.
[31]Webb TE,Poulter M,Beck J,et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series[J]. Brain:a Journal of Neurology,2008,131(Pt 10):2632-26346.
[32]Arata H,Takashima H,Hirano R,et al. Early clinical signs and imaging findings in Gerstmann-Straussler-Scheinker syndrome (Pro102Leu)[J]. Neurology,2006,66(11):1672-1678.
[33]Takazawa T,Ikeda K,Ito H,et al. A Distinct Phenotype of Leg Hyperreflexia in a Japanese Family with Gerstmann-Sträussler-Scheinker Syndrome (P102L)[J]. Internal Medicine,2010,49(4):339-342.
[34]Yamada M,Tomimitsu H,Yokota T,et al. Involvement of the spinal posterior horn in Gerstmann-Straussler-Scheinker disease (PrP P102L)[J]. Neurology,1999,52(2):260-265.
[35]Irisawa M,Amanuma M,Kozawa E,et al. A case of Gerstmann-Straussler-Scheinker syndrome. [J]. Mrms,2007,6(1):53-57.
[36]Konaka K,Kaido M,Okuda Y,et al. Proton magnetic resonance spectroscopy of a patient with Gerstmann-Straussler-Scheinker disease[J]. Neuroradiology,2000,42(9):662-665.
[37]Cagnoli C,Brussino A,Sbaiz L,et al. A previously undiagnosed case of Gerstmann-Straussler-Scheinker disease revealed by PRNP gene analysis in patients with adult-onset ataxia[J]. Movement Disorders:Official Journal of the Movement Disorder Society,2008,23(10):1468-1471.
[38]Woulfe J,Kertesz A,Frohn I,et al. Gerstmann-Straussler-Scheinker disease with the Q217R mutation mimicking frontotemporal dementia[J]. Acta Neuropathologica,2005,110(3):317-319.
[39]Karmon Y,Kurzweil A,Lindzen E,et al. Gerstmann-Straussler-Scheinker syndrome masquerading as multiple sclerosis[J]. Journal of the Neurological Sciences,2011,309(1-2):55-57.
1003-2754(2016)11-1032-03
R742.8+9
短篇与个案报告
2016-09-10;
2016-10-16
(吉林大学白求恩第一医院神经内科和神经科学中心,吉林 长春 130021)
冯加纯,E-mail:fengjcfrank@126.com
