大豆皂苷Ⅰ抑制唾液酸转移酶的分子机理研究
2015-11-20王棐张海玲光翠娥桑尚源杨红飞
王棐,张海玲,光翠娥,桑尚源,杨红飞
(食品科学与技术国家重点实验室,江南大学,江苏无锡 214122)
大豆皂苷Ⅰ抑制唾液酸转移酶的分子机理研究
王棐,张海玲,光翠娥*,桑尚源,杨红飞
(食品科学与技术国家重点实验室,江南大学,江苏无锡 214122)
从晶体结构出发,应用分子对接和结合自由能分析,研究了唾液酸转移酶(Sialyltransferase,ST)与其抑制剂大豆皂苷Ⅰ的相互作用机理,确定了它们的作用位点、作用力类型及大小。结果表明:范德华力和静电相互作用是复合物形成的主要驱动力,极性溶剂化能则起相反作用;8个氨基酸残基Gly149、Ser151、Met172、Asn173、Phe292、Trp300、His301、Ser325与大豆皂苷Ⅰ形成疏水相互作用,11个氨基酸残基Asn150、Tyr194、Ser271、Thr272、Gly273、Ile274、Gly291、Gly293、His302、Glu305、Glu324与大豆皂苷Ⅰ形成氢键作用;大豆皂苷Ⅰ占据了ST与底物胞苷一磷酸-β-N-乙酰神经氨酸相互作用的12个氨基酸残基中的11个,可起到竞争性抑制的作用。
唾液酸转移酶;大豆皂苷Ⅰ;分子对接;结合自由能分析
唾液酸转移酶(ST)是一类糖基转移酶,它以胞苷一磷酸-β-N-乙酰神经氨酸(CMP-β-N-acetylneuramic acid,CMP-Neu5Ac)为底物,将唾液酸残基Neu5Ac以α-2,3,α-2,6或α-2,8糖苷键的形式转移至新的糖基受体上形成唾液酸糖苷化合物[1]。细胞表面糖蛋白和糖脂的唾液酸化修饰在许多生物过程中发挥着至关重要的作用,如致癌性转化、肿瘤转移和入侵[2]。
ST的底物单一,唾液酸供体仅为CMP-Neu5Ac[1],所以,迄今为止,ST抑制剂的设计主要是基于供体CMP-Neu5Ac的化学结构,现在也有研究是基于CMP-Neu5Ac过渡态的结构和糖基受体的结构进行设计以及通过随机筛选得到[3]。然而,由于低膜通透性这些化合物很难进入细胞或组织[3]。Chi-Yue Wu[2]等人发现大豆皂苷Ⅰ对ST中的ST3Gal I有强烈的抑制作用,抑制常数(K1)为2.3 μmol/L,作为竞争性抑制剂(相对底物CMP-Neu5Ac),大豆皂苷Ⅰ对ST3Gal I的亲和性是CMP-Neu5Ac和CMP的20和25倍,并且大豆皂苷Ⅰ只对ST起作用,对其他的糖基转移酶和糖苷酶几乎不起抑制作用。细胞水平上,大豆皂苷Ⅰ能抑制高转移黑色素瘤细胞表面唾液酸的生成[4],也能抑制MFC-7乳腺癌细胞ST的活性、ST3GalⅣmRNA的表达及唾液酸的分泌[5]。
ST是药物设计的重要靶点,作者采用分子对接及结合自由能计算的方法研究大豆皂苷Ⅰ对ST的抑制作用,确定他们的作用力类型、结合的关键位点以及复合物的结合自由能,提高了实验效率,减少不必要的消耗,为ST抑制剂的合理药物设计提供理论依据,也为大豆皂苷Ⅰ的开发利用提供了新思路。
1 材料与方法
1.1 受体ST与配体大豆皂苷Ⅰ分子的获取
ST的三维结构取自蛋白质晶体结构数据库(http://www.rcsb.org/pdb/),PDB ID:2WNB,分辨率为0.155 nm,共有298个氨基酸残基,相对分子质量为34 748.3,小分子配体为C8H15NO6、C9H14N3O8P、C6H6NO3和C6H12O6,利用北京创腾科技有限公司的Discovery Studio 2.5(DS)软件去掉小分子配体和水分子并加氢后得到ST三维结构并作为受体用于后续的对接过程。大豆皂苷Ⅰ(图1)的三维结构取自chemicalbook网站库(http://www.chemicalbook.com),分子式为C48H78O18,相对分子质量为943.12,采用PRODRG网站(http://davapc1.bioch.dundee.ac.uk/cgibin/prodrg)对大豆皂苷Ⅰ的分子结构进行加电荷和构象优化等预处理。
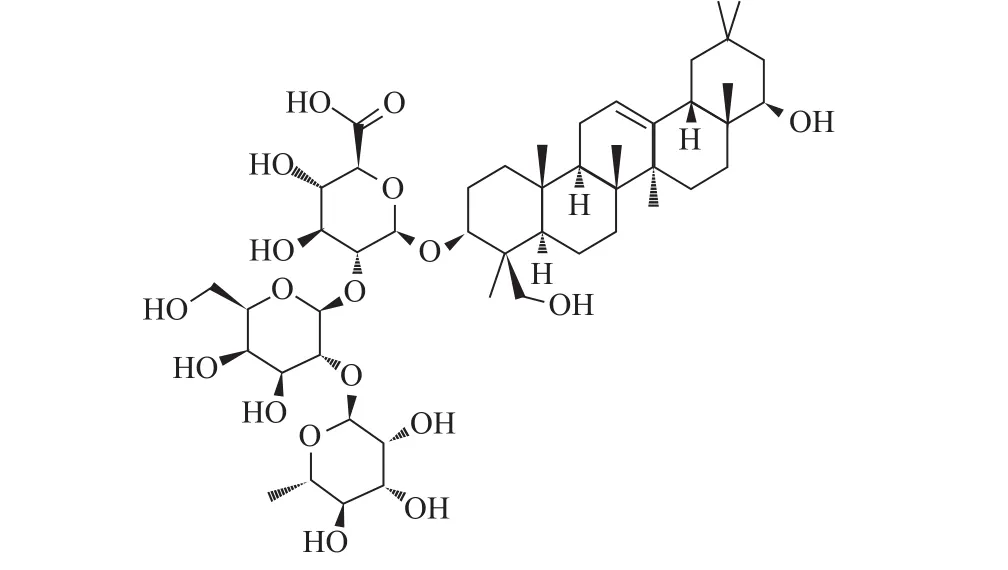
图1 大豆皂苷Ⅰ的分子结构Fig.1Molecular structure of soyasaponinⅠ
1.2 ST与大豆皂苷Ⅰ的分子对接
以ST为对接受体,以大豆皂苷Ⅰ为对接配体,以ST的活性位点Gln108、Asn150、Met172、Asn173、Tyr194、Clu196、Phe212、Tyr233、Arg269、Thr272、Gly273、Gly293、Trp300、His302、Val318、His319形成的区域为中心,通过DS软件包中的Libdock模块,在半径为1.65 nm内的范围内(图2)进行受体与配体的分子对接。每产生一个构象就进行一次分子对接,配体分子在受体口袋中可能的构象模式采用Libdock模块进行收集。根据LibDock综合得分LibDockscore和最终的均方根偏差RMSD值,分析和评估ST与大豆皂苷Ⅰ的相互作用。取打分最好以及底物骨架重合性较高的复合物构象,运用DS程序包,采用CHARMm力场,添加抗衡离子溶剂化(Solvation)后,对对接复合物的空间结构进行两次能量优化,electrostatic参数设置为Particle,Mesn,Ewald。优化分成2步完成:先限制蛋白质和抑制剂的结构,只优化水分子;再去除限制,优化整个体系。每个步骤都先采用最陡下降法(Steepest Descent)优化500步,再采用共轭梯度法(Conjugate Gradient)优化1 000步。基于优化后的空间结构用Ligplot+软件统计受体中与配体产生氢键及疏水作用的氨基酸残基,并分析复合物的空间作用力。
1.3 对接复合物的结合自由能计算
基于优化后的结构,用分子力学泊松-波尔兹曼表面积法(molecular mechanics Poisson-Boltzmann surface area,MM-PBSA)计算对接复合物的结合自由能(binding free energy,ΔGbind)[6]。ΔGbind值越低说明受体与配体之间的亲和力越高[7]。MM-PBSA法将体系自由能分解为真空下分子内能、溶剂化能和由构象变化引起的熵变3部分,能更为直观的分析各种相互作用的贡献。结合自由能(ΔGbind)计算过程为:
图2 ST与大豆皂苷Ⅰ分子对接的活性区域图Fig.2Active zone of ST docking with soyasaponinⅠ

其中Gcomplex、Greceptor和Gligand分别为复合物、受体和配体的自由能。复合物、受体或配体的自由能由液相和气相两部分组成的,液相部分的自由能为溶剂化能Gsolv,主要包括极性溶剂化能GPB和非极性溶剂化能GSUR,用DS中的程序包计算,Implicit Solvent Model参数设置为Poisson Boltzmann with non-polar Surface Area,electrostatic参数设置为Spherical Cutoff;气相部分的自由能包含内能EMM和熵TSMM,EMM为整个分子力学能,Eelec和EvdW分别为气相中分子静电相互作用和范德华能,用DS中的程序包计算,Implicit Solvent Model参数设置为None,electrostatic参数设置为Spherical Cutoff。因为TSMM值的变化对结合自由能的影响非常小,所以一般情况下可以忽略此数值。
2 结果与讨论
2.1 分子对接总体结果
从晶体结构看出,ST3Gal-I按结构折叠方式属于GTA类,有12个α螺旋,7个β折叠,3个二硫键(Cys62-Cys67、Cys65-Cys142、Cys145-Cys284)[8]。Libdock模块收集的大豆皂苷Ⅰ与ST活性口袋结合的可能构象显示,配体与受体蛋白的结合方式共有90种,其中得分超过100的有85种,LibDock综合得分最高为186.2表明,大豆皂苷Ⅰ和ST活性区域匹配性较好(LibDockscore>100)。以打分最高的对接结果为研究对象(图3,(a)),将其与结合有糖基受体Galβ1,3GalNAcα-PhNO2和产物CMP的ST3Gal-I(图3,(b))、结合有糖基受体Galβ1,3GalNAcα-PhNO2和底物CMP-Neu5Ac的ST3Gal-I(图3,(c))、结合有糖基受体Galβ1,3GalNAcα-PhNO2和大豆皂苷Ⅰ的ST3Gal-I进行比较(图3,(d)),结果表明大豆皂苷Ⅰ主要与ST中由氨基酸残基Asn150、Met172、Asn173、Gly273、Gly293、Trp300和His302形成的活性区域产生相互作用,这类似于CMP-Neu5Ac与ST的作用,并且发现糖基受体的存在几乎不影响大豆皂苷Ⅰ和ST活性区域的结合。

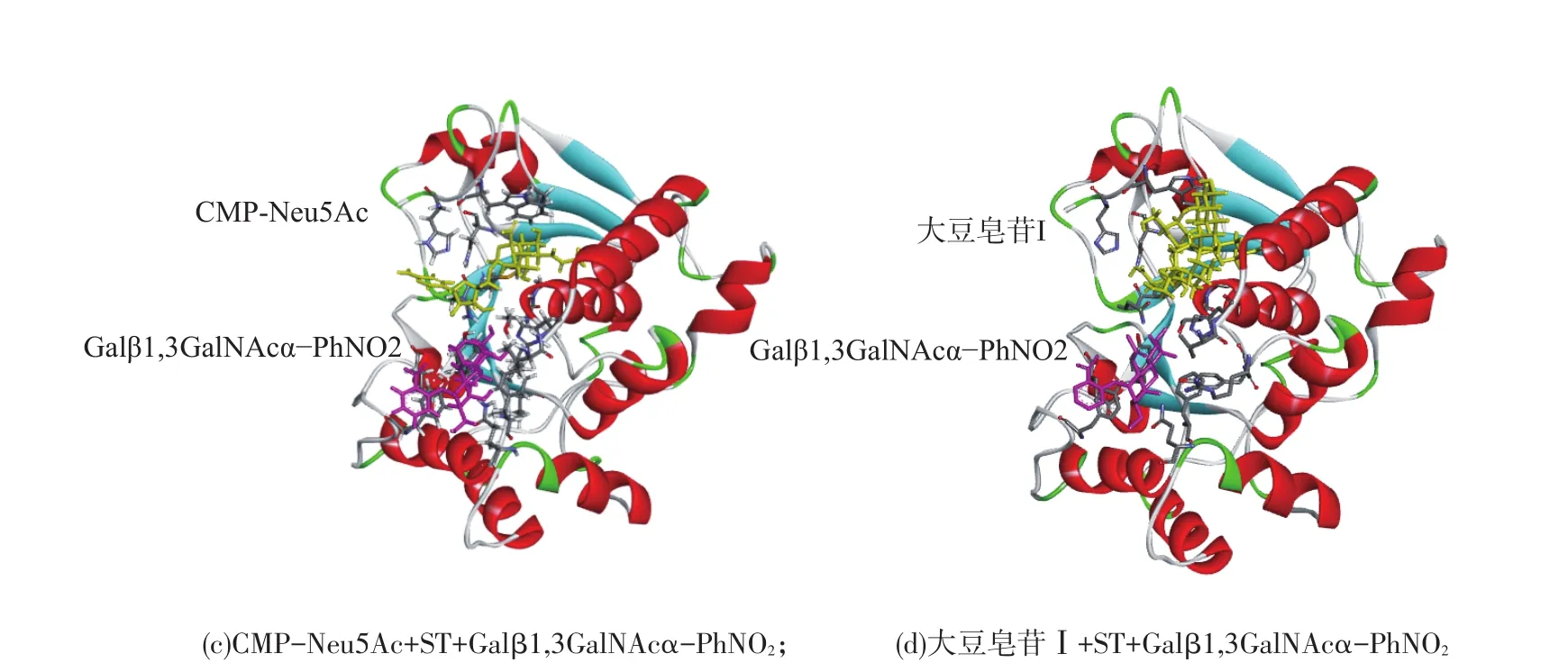
图3 ST与配体分子对接示意Fig.3Molecular docking schematic diagram of ST andligands
2.2 大豆皂苷Ⅰ与ST形成的氢键和疏水作用分析
图4为用Ligplot+软件显示的对接后配体与受体相互作用的二维图。

图4 ST和大豆皂苷Ⅰ分子间氢键和疏水相互作用分析Fig.4Hydrogen bond and hydrophobic interactions between ST and soyasaponinⅠ
通过图4分析可得,ST中8个氨基酸残基Gly149、Ser151、Met172、Asn173、Phe292、Trp300、His301、Ser325与大豆皂苷Ⅰ形成疏水相互作用,11个氨基酸残基参与形成氢键作用(表1)。其中氨基酸Asn150、Gly273、Gly293也是CMP-Neu5Ac及CMP与ST形成氢键的重要残基[8],Ile274、Gly291、Glu305也可影响CMP与ST的作用,Thr194、Ser271、Ser272也可影响Gal与ST的作用。ST与大豆皂苷Ⅰ的氢键和疏水作用结果表明,大豆皂苷Ⅰ能有效阻碍CMP-Neu5Ac与ST的作用。

表1 ST和大豆皂苷I的氢键分析Table 1Hydrogen bondsbetween ST and soyasaponin I
2.3 复合物的比较
通过Ligplot+软件将大豆皂苷Ⅰ以及CMPNeu5Ac分别与ST的对接结果进行比较[9](图5),发现CMP-Neu5Ac与ST的12个氨基酸残基存在相互作用,其中11个氨基酸残基Asn150、Ser151、Met172、Asn173、Tyr194、Thr272、Gly273、Phe292、Gly293、Trp300、His301在ST与大豆皂苷Ⅰ、ST与CMPNeu5Ac的结合过程中都参与了作用,并且CMPNeu5Ac与ST分子对接的LibDock综合得分最高为143.5,得分超过100的结合方式有73种,说明大豆皂苷Ⅰ能有效阻止CMP-Neu5Ac与ST的结合。通过图6看出,大豆皂苷Ⅰ在空间位置上也能阻碍CMP-Neu5Ac与ST的结合。

图5 大豆皂苷I-ST与CMP-Neu5Ac-ST相互作用细节比对Fig.5Interaction comparison between soyasaponin IST and CMP-Neu5Ac-ST

图6 大豆皂苷I与CMP-Neu5Ac在ST活性口袋中的空间位置Fig.6Spatial superposition of soyasaponin I and CMP-Neu5Ac in the active site of ST
2.4 大豆皂苷Ⅰ与ST复合物的结合自由能计算结果
结合自由能反映了受体和配体结合的稳定性[10]。对大豆皂苷Ⅰ和ST进行分子对接和优化后,按照公式(1)对对接复合物的结合自由能进行计算。结果发现大豆皂苷Ⅰ和ST的结合自由能(ΔGbind)为-209 kJ/mol,表明大豆皂苷Ⅰ和ST结合稳定。

图7 结合自由能各项能量示意图Fig.7Each energy contributiong value in binding free energy
通过对比图7中结合自由能各项能量的贡献值可以看出,在大豆皂苷Ⅰ和ST形成的复合物中,ΔEvdW,ΔEelec和ΔGSUR均为负值,表明范德华力、静电相互作用和非极性溶剂化能对结合自由能都起到促进作用,而极性溶剂化能很大程度上表现为阻碍ST和大豆皂苷Ⅰ的结合。ΔEvdW和ΔEelec在ΔGbind中占有主导地位,表明范德华力和静电相互作用对于大豆皂苷Ⅰ和ST之间的结合能作用贡献比较大(>90%),但是较强的极性溶剂化能可抵消部分作用力,阻碍大豆皂苷Ⅰ和ST的结合。
3 结语
对大豆皂苷I与ST进行分子对接,预测了ST与大豆皂苷I相互作用的模式,得到的复合物结果表明:8个氨基酸残基与大豆皂苷I形成疏水相互作用,11个氨基酸残基与大豆皂苷I形成氢键作用;结合自由能为-209 kJ/mol,范德华力和静电相互作用是复合物形成的主要驱动力,极性溶剂化能不利于复合物的形成;大豆皂苷I相对底物是ST的竞争性抑制剂。
[1]王亚娟,邢国文.唾液酸酶和唾液酸糖基转移酶的结构,功能与催化反应研究进展[J].有机化学,2011,31(8):1157-1168.
WANG Yajuan,XING Guowen.Progress in structure,function and catalytic reactions of sialidase and sialyltransferase[J].Chinese Journal of Organic Chemistry,2011,31(8):1157-1168
[2]Wu C Y,Hsu C C,Chen S T,et al.Soyasaponin I,a potent and specific sialyltransferase inhibitor[J].Biochemical and biophysical research communications,2001,284(2):466-469.
[3]Wang X,Zhang L H,Ye X S.Recent development in the design of sialyltransferaseinhibitors[J].Medicinal research reviews,2003,23(1):32-47.
[4]Chang W W,Yu C Y,Lin T W,et al.Soyasaponin I decreases the expression of α2,3-linked sialic acid on the cell surface and suppresses the metastatic potential of B16F10 melanoma cells[J].Biochemical and biophysical research communications,2006,341(2):614-619.
[5]Hsu C C,Lin T W,Chang W W,et al.Soyasaponin-I-modified invasive behavior of cancer by changing cell surface sialic acids[J]. Gynecologic oncology,2005,96(2):415-422.
[6]Zhang Y,Pan D,Shen Y,et al.Understanding the molecular mechanism of the broad and potent neutralization of HIV-1 by antibody VRC01 from the perspective of molecular dynamics simulation and binding free energy calculations[J].Journal of molecular modeling,2012,18(9):4517-4527
[7]Takamatsu Y,Sugiyama A,Purqon A,et al.Binding free energy calculation and structural analysis for antigen-antibody complex[C]//AIP Conference Proceedings.2006,832(5):566-569.
[8]Rao F V,Rich J R,Rakic B,et al.Structural insight into mammalian sialyltransferases[J].Nature Structural and Molecular Biology,2009,16(11):1186.
[9]Laskowski RA,Swindells MB.LigPlot+:multiple ligand-protein interaction diagrams for drug discovery[J].Journal of chemical information and modeling,2011,51:2778-2786.
[10]Guang C,Shang J,Jiang B.Transport of traditional Chinese pimple milk-derived angiotensin-converting enzyme(ACE)inhibitory peptides across a Caco-2 cell monolayer and their molecular recognition with ACE[J].Journal of Food,Agriculture& Environment,2012,10(3&4):40-44.
Mechanism of Interaction between Sialyltransferase and Its Inhibitory SoyasaponinⅠ
WANG Fei,ZHANG Hailing,GUANG Cuie*,SANG Shangyuan,YANG Hongfei
(State Key Laboratory of Food Science and Technology,Jiangnan University,Wuxi 214122,China)
The molecular mechanism for the binding sites,driving forces and the interaction intensity between sialyltransferase(ST)and soyasaponin I was investigated by molecular docking and free energy calculation based on the crystal structure.The van der Waals force and electrostatic interaction were considered as the main driving forces,while polar solvation energy behaved in an opposite manner.The hydrophobic interaction was formed between soyasaponin I and eight amino acid residues,i.e.,Gly149,Ser151,Met172,Asn173,Phe292,Trp300,His301,and Ser325.Hydrogen bonding was found between soyasaponin I and eleven amino acid residues,including Asn150,Tyr194,Ser271,Thr272,Gly273,Ile274,Gly291,Gly293,His302,Glu305,and Glu324.Soyasaponin I was a competitive inhibitor which occupied 11 of the 12 amino acid residues that accommodated the substrate CMP-β-N-acetylneuramic acid.These results provide a theoretical basis for the development and utilization of soyasaponin I as a ST inhibitor.
sialyltransferase,soyasaponin I,molecular docking,free energy calculation
R914.2
A
1673—1689(2015)04—0355—06
2014-07-15
国家自然科学基金项目(31201289);江南大学大学生创新训练计划项目(012011342)。
*通讯作者:光翠娥(1976-),女,湖北仙桃人,工学博士,副教授,主要从事食品营养与功能因子研究。E-mail:guang1226@hotmail.com。
