基于支持向量机的蛋白质相互作用界面热点残基预测
2015-08-09天津科技大学计算机科学与信息工程学院天津300222天津瑞和天孚科技有限公司天津300384
(1. 天津科技大学计算机科学与信息工程学院,天津 300222;2. 天津瑞和天孚科技有限公司,天津 300384)
(1. 天津科技大学计算机科学与信息工程学院,天津 300222;2. 天津瑞和天孚科技有限公司,天津 300384)
针对蛋白质相互作用界面中的热点残基是局部紧凑地聚集着,而现有的基于机器学习的热点残基预测方法仅从目标残基中提取特征,并没有考虑目标残基的局部空间结构信息,以及如何进行特征提取并获得非冗余的特征子集等问题,为准确识别蛋白质相互作用界面的热点残基,提出结合蛋白质相互作用界面残基的空间邻近残基信息提取多类特征,并利用随机森林来进行特征提取,最后利用支持向量机来预测热点残基的方法.计算实验表明,该预测方法可以有效地用来发现热点残基.
蛋白质相互作用界面;热点;支持向量机;随机森林
蛋白质经常通过蛋白质间相互作用来行使其功能,例如信号传导网络和代谢网络中的蛋白质复合物,而蛋白质相互作用界面是蛋白质相互作用发生的物理载体.实验证明蛋白质相互作用界面上残基的结合能量并不是均匀分布的,而是一些残基的结合能量较大而且仅占界面残基的一小部分,这些对于蛋白质结合起关键作用的残基称为热点(hot spots)[1].丙氨酸扫描变异(Alanine scanning mutagenesis)是目前主要的识别热点的实验方法,其基本原理是把界面上的单个残基替换成丙氨酸,并测得替换以后残基结合能量的变化值.选择丙氨酸作为替换残基是因为丙氨酸的侧链仅有一个碳原子,并且替换后不改变主链构象,也不会产生很大的静电或者位阻效应[2].由于其实验过程较为复杂,目前获得的丙氨酸扫描变异数据很少,主要存放在丙氨酸扫描变异数据库ASEdb[3]和结合界面残基数据库 BID[4]中.目前,已经有一些研究工作来刻画热点残基的序列和结构特点,例如:分析热点残基和非热点残基的氨基酸组成,发现色氨酸、精氨酸和酪氨酸更易形成热点残基,而亮氨酸、丝氨酸、苏氨酸和缬氨酸更易形成非热点残基[5];O环理论认为蛋白质相互作用界面的热点被对结合能量贡献不大的残基形成环并包裹着,这些形成环的残基用来隔离热点残基和水分子[6].
基于已有的对热点残基的序列和结构特点的研究,目前有一些基于机器学习的方法来预测蛋白质相互作用界面热点,并取得了相对较高的预测精度[7],但是相关研究领域仍有一些问题存在,具体表现为:(1)蛋白质相互作用界面中的热点残基被发现是局部紧凑地聚集着,而现有的热点残基预测方法仅从目标残基中提取特征并用来训练分类器,如何有效地利用目标残基的局部空间结构信息来提高预测精度是需要考虑的;(2)尽管目前已经提出了许多分类特征,如何进行特征提取并获得非冗余的分类特征也是需要考虑的.
本文从目标残基及它的2个空间相邻残基,即相互作用界面另一侧的距离最近的残基(镜面接触残基)和同一侧的距离最近的残基(内部接触残基),来获取分类特征;然后结合随机森林来估计分类特征的重要性,并进行特征提取;最后利用支持向量机来有效地整合特征并用于热点残基预测.
1 数据获取
首先从丙氨酸扫描变异数据库(ASEdb)中获取含有丙氨酸扫描变异残基的蛋白质链及相关复合物.对于蛋白质相互作用界面残基,当其结合能量的变化值(ΔΔG)≥8.364,kJ/mol时,定义该残基为热点[7].这样,训练集包括来自20个蛋白质复合物中的318个丙氨酸扫描变异残基,其中 77个残基是热点残基,241个残基是非热点残基.另外,利用BID中的数据集作为独立测试集,包括 18个蛋白质复合物中的125个界面残基,其中 38个残基是热点残基,87个残基是非热点残基.关于训练集和测试集的详细描述参见文献[7].
2 计算方法
2.1 分类特征描述
对于蛋白质相互作用界面残基,本文设计了多个分类特征描述符,用于热点预测和分类,并且基于它们的不同来源和性质,将其大体分为5类[7].
2.1.1 原子接触数和原子接触面积
对于2个残基中的各自1个原子,通过CSU程序[8]定义它们的接触关系(contact atoms),其是基于原子间的距离以及所在环境的拥挤程度来确定的.进而,对于 1个残基i,通过对残基i与相互作用界面中其他残基j的接触原子数目求和来定义残基i的原子接触数.另外,通过对相互作用界面另一侧残基 j的原子接触面积求和来定义残基i的原子接触面积.
2.1.2 残基接触数和物理化学特征
2个残基中如果至少有 1对接触原子(2个原子分别来自于2个残基),则这2个残基称为接触残基(contact residues).对于残基i,利用相互作用界面中的接触残基 j的数目定义残基i的残基接触数.另外,考虑残基i的6个物理化学特征(包括疏水性、亲水性、等电点、质量、极性和极化率),其中i的每个物理化学特征通过对所有接触残基j的相应物理化学参数求和以定义残基i的物理化学特征.
2.1.3 相对可及表面积和相对侧链可及表面积
可及表面积是指生物分子对于溶剂的可接触表面积,残基的可及表面积与蛋白质的功能和活性位点有密切关系.这里残基的相对可及表面积和相对侧链可及表面积分别度量了残基和侧链在形成蛋白质复合物后的可及表面积的变化率.
2.1.4 深度指数
原子的深度定义为该原子和最近的溶剂可及原子之间的距离.这里通过 PSAIA程序[9]计算残基的以下特征描述符:平均深度指数(残基所有原子的平均深度指数)、深度指数的标准差、侧链平均深度指数(侧链所有原子的平均深度指数)、侧链深度指数的标准差.另外,本文还计算了残基和侧链的相对深度指数(分别为残基和侧链在形成蛋白质复合物后的平均深度指数的变化率).
2.1.5 二级结构和氨基酸分类
残基的二级结构包括螺旋、折叠或卷曲.另外,基于偶极矩与侧链体积,20种蛋白质氨基酸被分为6类,第 1类:天冬氨酸、谷氨酸;第 2类:精氨酸、赖氨酸;第 3类:丙氨酸、甘氨酸、缬氨酸;第 4类:酪氨酸、甲硫氨酸、苏氨酸、丝氨酸、半胱氨酸;第 5类:异亮氨酸、亮氨酸、苯丙氨酸、脯氨酸;第 6类:组氨酸、天冬酰胺、谷氨酰胺、色氨酸.因此,这部分包括2个离散特征描述符,其变量取值个数分别为3和6.
基于上面5类特征,对于1个残基共有19个特征描述符.为了考虑目标残基的空间结构信息,本研究从目标残基、镜面接触残基和内部接触残基获取分类特征描述符,并作为目标残基的特征.这样对于 1个目标残基,获取的特征个数为57.
2.2 特征选择
特征选择是训练分类器前的重要一步,并且其通过去掉冗余和不相关的特征,提高分类器的预测性能.在这里,对目标残基共提出了57个特征,这样的特征集可能会引起模型的过拟合,因此,使用随机森林挑选出重要的特征,以便更好地区别热点残基和非热点残基.
随机森林是包含多个决策树的分类器,并且其输出的类别是由个别树输出的类别的众数而定.在决定类别的同时,随机森林还提供了评估变量重要性的方法,其中最常用的是基于袋外数据(OOB)的特征值随机扰动后,度量其袋外数据分类精度的平均下降值.利用该度量方法进行特征选择,并通过R软件包randomForest进行计算.
2.3 分类算法
支持向量机是一种监督式学习的方法,广泛地应用于统计分类以及回归分析.支持向量机将向量映射到更高维的空间里,在这个空间里建立有1个最大间隔超平面.在分开数据的超平面的两边建有 2个互相平行的超平面,分隔超平面使2个平行超平面的距离最大化.这里通过 R软件包 e1071建立支持向量机分类器.
2.4 预测性能的度量
为了度量所提热点预测方法的分类性能,本文采用一些常用的指标,包括预测精度(nACC)、敏感性(nSE)、准确率(nPR)、特异性(nSP)和 Matthew相关系数(nMCC).这些指标的具体定义如下:

式中 nTP、nFP、nTN和 nFN分别表示真正类的数量(正确预测的热点残基)、假正类的数量(非热点残基被错误地预测为热点残基)、真负类的数量(正确预测的非热点残基)和假负类的数量(热点残基被错误地预测为非热点残基).
ROC曲线是用构图法揭示敏感性与特异性的相互关系,曲线本身以及相伴随的指标——曲线下面积(nAUC)常被用来度量分类器的预测性能,nAUC值越接近于1,说明分类效果越好.
3 计算结果与讨论
3.1 估计特征的重要性
利用随机森林估计初始 57个特征的重要性.表1给出了前 16个重要特征,是基于袋外数据分类精度的平均下降值排序的.
针对表1中的16个重要特征,对于目标残基和镜面接触残基,依据 2.1节对特征描述符的分类,从每类特征描述符中选取1个最重要特征(利用表1衡量特征的重要性),最终选择了7个特征(目标残基的原子接触面积、目标残基的质量、镜面接触残基的残基接触数、目标残基的相对侧链可及表面积、镜面接触残基的相对侧链可及表面积、目标残基的侧链平均深度指数、镜面接触残基的平均深度指数),用于支持向量机分类器的建立.
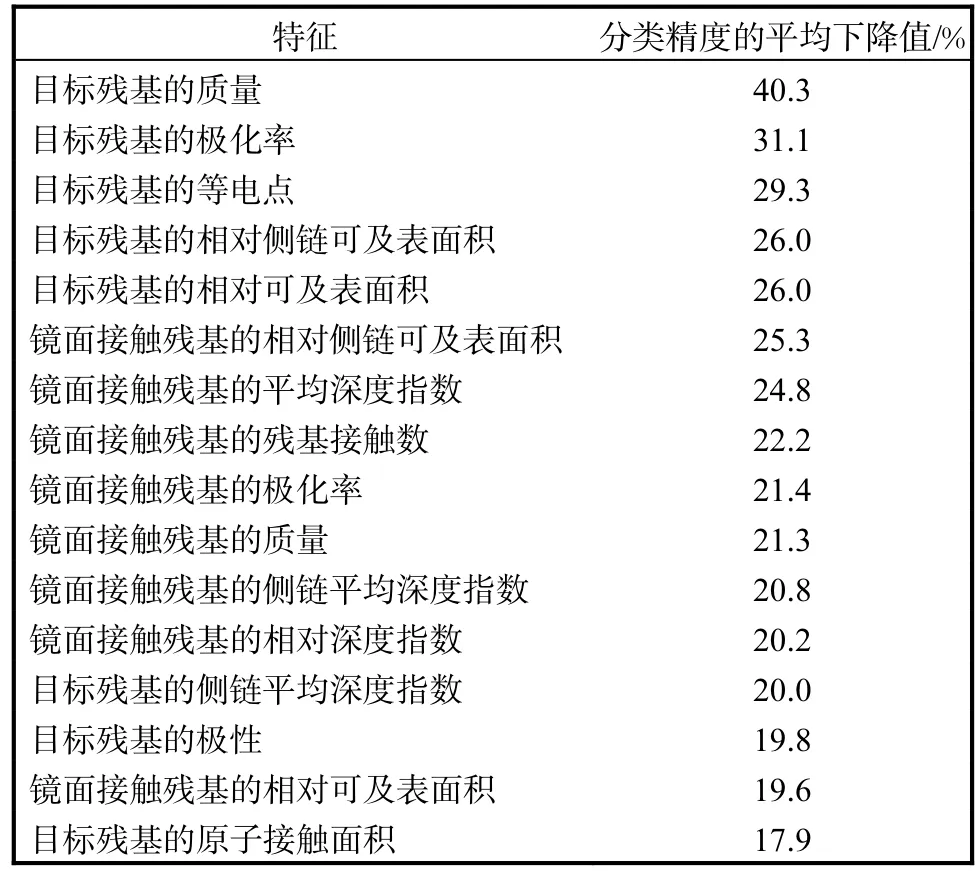
表1 利用随机森林估计的前16个重要特征Tab. 1 The first 16 important characteristics evaluated by random forests
3.2 基于训练集的5折交叉验证
在训练集上通过 5折交叉验证检验基于支持向量机的分类器的预测性能.数据集被随机分成样本数量近似相等的 5份,然后依次选择每 1份为测试集,剩下的 4份为训练集,建立分类器.基于该计算过程,预测精度nACC=84.0%,敏感性nSE=46.8%,准确率 nPR=78.3%,特异性 nSP=95.9%,Matthew相关系数nMCC=0.519.另外,图1给出了分类器的ROC曲线,其曲线下面积 nAUC=0.762.这些预测结果显示:采用所选特征,利用基于支持向量机方法能够有效地区分热点残基和非热点残基.

图1 支持向量机分类器的ROC曲线Fig. 1 ROC curve of support vector machine classifier
为了进一步考察各类物理量对于热点预测性能的影响,依次删除不同物理量后,同样采用 5折交叉验证的方法计算 ROC曲线下面积 nAUC,结果见表2.可以看出,删除不同物理量后,nAUC值均有所减小,所以这些物理量都有助于热点预测性能的提高.

表2 依次删除不同物理量后在训练集上的预测性能比较Tab. 2 Comparison of predicting performance in the training set after subtracting each physical quantity
3.3 独立测试集上的预测性能
在独立测试集上比较所提方法和已有热点预测方法的预测性能.现有的热点预测方法主要包括基于能量的方法 Robetta[10]和 FOLDEF[11]、基于决策树的方法 KFC[12]以及经验方法 HotPoint[13].表 3给出了不同方法的预测性能,其中这些比较方法的预测结果是通过它们各自的网页服务器计算获得的.本文基于支持向量机的预测方法的预测结果为nPR=60.0%,nSE=31.6%,nSP=90.8%,nACC=72.8%,nMCC= 0.281.从表 3可以看出,本文方法在准确率、特异性和预测精度方面要优于其他热点预测方法,并且相对于其他预测方法的最好结果,其分别提高了 8%,3.4%和2.4%.
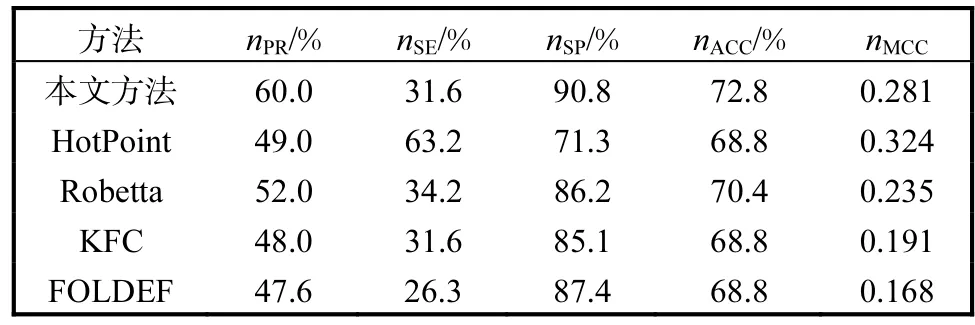
表3 不同热点预测方法在测试集上的性能比较Tab. 3 Comparison of different hot spot predicting methods in the test set
4 结 语
本文提出了一种新的计算方法以识别蛋白质相互作用界面的热点,即从目标残基、镜面接触残基和内部接触残基获取各类特征,并且利用随机森林选择重要特征,最后利用支持向量机有效整合这些特征.计算结果表明,该方法可以有效地用于热点预测.文中计算用的数据集和代码可从以下网址下载:http://sourceforge.net/projects/tustbioinfor/files/.
[1] Bogan A A,Thorn K S. Anatomy of hot spots in protein interfaces[J]. Journal of Molecular Biology,1998,280(1):1-9.
[2] Cunningham B C,Wells J A. High-resolution epitope mapping of hgh-receptor interaction by alanine-scanning mutagenesis[J]. Science,1989,244(4908):1081-1085.
[3] Thorn K S,Bogan A A. ASEdb:A database of alanine mutations and their effects on the free energy of binding in protein interactions[J]. Bioinformatics,2001,17(3):284-285.
[4] Fischer T B,Arunachalam K V,Bailey D,et al. The binding interface database(BID):A compilation of amino acid hot spots in protein interfaces[J]. Bioinformatics,2003,19(11):1453-1454.
[5] Moreira I S,Femandes P A,Ramos M J. Hot spots-A review of the protein-protein interface determinant amino-acid residues[J]. Proteins,2007,68(4):803-812.
[6] Li X,Keskin O,Ma B,et al. Protein-protein interactions:Hot spots and structurally conserved residues often locate in complemented pockets that preorganized in the unbound states:Implications for docking[J]. Journal of Molecular Biology,2004,344(3):781-795.
[7] Wang L,Liu Z P,Zhang X S,et al. Prediction of hot spots in protein interfaces using a random forest model with hybrid features[J]. Protein Engineering Design and Selection,2012,25(3):119-126.
[8] Sobolev V,Sorokine A,Prilusky J,et al. Automated analysis of interatomic contacts in proteins[J]. Bioinformatics,1999,15(4):327-332.
[9] Mihel J,Sikic M,Tomic S,et al. PSAIA-protein structure and interaction analyzer[J]. BMC Structural Biology,2008,8(1):21.
[10] Kortemme T,Baker D. A simple physical model for bind ing energy hot spots in protein-protein complexes[J]. Proceedings of the National Academy of Sciences of the United States of America,2002,99(22):14116-14121.
[11] Guerois R,Nielsen J E,Serrano L. Predicting changes in the stability of proteins and protein complexes:A study of more than 1000 mutations[J]. Journal of Molecular Biology,2002,320(2):369-387.
[12] Darnell S,Page D,Mitchell J C. An automated decisiontree approach to predicting protein interaction hot spots[J]. Proteins,2007,68(4):813-823.
[13] Tuncbag N,Gursoy A,Keskin O. Identification of computational hot spots in protein interfaces:Combining solvent accessibility and inter-residue potentials improves the accuracy[J]. Bioinformatics,2009,25(12):1513-1520.
基于支持向量机的蛋白质相互作用界面热点残基预测
畅卫功1,李 灏2,王 林1,杨海波1
Predicting of Hot Spots at Protein Interfaces Using Support Vector Machines
CHANG Weigong1,LI Hao2,WANG Lin1,YANG Haibo1
(1. College of Computer Science and Information Engineering,Tianjin University of Science & Technology,Tianjin 300222,China;2. Tianjin Rui He Tian Fu Science & Technology Ltd. Co.,Tianjin 300384,China)
Hot spots at protein interfaces were found to be clustered within locally and tightly packed regions. However,the existing machine learning based on hot spot prediction methods only gets features from the target residue,and does not consider the local spatial information of the target residue. Meanwhile,how to conduct the feature selection and obtain the subsets without redundant features should also be considered. In order to accurately identify hot spot residues at protein interfaces,this research tried to get various features by taking into consideration the spatial neighbor residues of each interface residue,and the feature selection was conducted by using random forests. Thereafter,the support vector machine was employed to predict the hot spots at protein interfaces. Computational experiments show that our prediction method can effectively discover hot spot residues.
protein interface;hot spot;support vector machine;random forest
TP399;Q816 文献标志码:A 文章编号:1672-6510(2015)02-0070-05
10.13364/j.issn.1672-6510.20140075
2014-05-14;
2014-08-28
天津市高等学校科技发展基金资助项目(20120803);天津市科技支撑计划重点资助项目(12ZCZDGX02400)
畅卫功(1974—),男,山西人,讲师,wgchang@tust.edu.cn.
常涛
