基于脂肪酸生物标记与16S rRNA的芽胞杆菌系统发育分析比较
2015-10-26刘国红刘波林营志唐建阳
刘国红 刘波 林营志 唐建阳
(福建省农业科学院农业生物资源研究所,福州 350003)
基于脂肪酸生物标记与16S rRNA的芽胞杆菌系统发育分析比较
刘国红 刘波 林营志 唐建阳
(福建省农业科学院农业生物资源研究所,福州 350003)
旨在探究脂肪酸作为一种有效的芽胞杆菌分类标记,以25种芽胞杆菌模式菌株为研究对象,对芽胞杆菌进行脂肪酸组分和16S rRNA基因系统进化分析比较。结果表明,脂肪酸系统发育分析能充分体现芽胞杆菌种类间的亲缘关系,并且按生物学特性进行聚类分群,而16S rRNA系统发育仅完美体现出种间的亲缘关系。利用脂肪酸分析可将25种芽胞杆菌完全准确分开,且将生物学特性相同的芽胞杆菌种类聚为一类,如碱性条件下生长良好的4种芽胞杆菌(B. agaradhaerens、B. alacalphilus、B. alkalitelluris和B. fastidiosus)聚为一类,芽胞杆菌为圆形的芽胞杆菌(B. fusiformis、B. odysseyi 和B. sphaericus)聚为一类。结果表明,脂肪酸分析不仅根据亲缘关系进行聚类,还可以根据生物学特性对芽胞杆菌进行分类。
芽胞杆菌;脂肪酸生物标记;16S rRNA;系统发育分析
芽胞杆菌属(Bacillus)属于细菌界(Bacteria),厚壁菌门(Firmicutes),杆菌纲(Bacilli),芽胞杆菌目(Bacillales),芽胞杆菌科(Bacillaceae),是一类好氧或兼性厌氧、产芽胞的革兰氏阳性杆状细菌。由于大多数芽胞杆菌种类可以产生多种多样的活性物质,在工业、农业、医学、环境等领域有着重要的经济价值,因此研究它们的分类地位对其应用有着更重要的研究意义[1,2]。
在研究微生物进化分类时,人们通常根据一些相对保守的序列进行分析,如rRNA序列、蛋白质的氨基酸序列、编码蛋白质的核酸序列。原因一方面是由于这些保守的序列会带来相对可靠的结果;另一方面是由于在数据库中,这些序列可以比较方便的得到。细菌分类及系统发育分析常借助16S rRNA作为标尺,但16S rRNA序列的保守性使得某些亲缘关系密切的种类无法区分开,在系统进化分析上存在一些缺陷[3]。鉴于芽胞杆菌分类的复杂性,使得寻找和建立这类细菌准确的分类方法颇受关注。由于细菌不同属、种,甚至不同株之间脂肪酸碳链长度、双键位置、取代基团等都存在差异,脂肪酸是细胞膜的重要遗传表达产物,与DNA具有同源性,因此脂肪酸分析技术在细菌分类中具有重要作用[4]。1963年,Able等[5]首次提出证据表明细胞脂肪酸可以成功的鉴定细菌。Kaneda[6]将22株芽胞杆菌分为6个群,Kämpfer[7]证明脂肪酸生物标记具有遗传稳定性,可以作为芽胞杆菌属种类分类鉴定的一种有效手段。张晓霞等[8]利用脂肪酸成分对不动杆菌进行鉴定,研究结果表明脂肪酸鉴定结果和16S rRNA基因分析结果一致,在种水平上利用16S rRNA基因系统发育分析结果与脂肪酸组分分析的结果可互为补充,相互印证。目前随着脂肪酸分析技术的日益成熟,基于脂肪酸分析技术的气相色谱Sherlock微生物鉴定系统,使脂肪酸分析更加快速、准确,在细菌分类中被广泛应用。脂肪酸分析可区分属、种并进行聚类。本研究对25种芽胞杆菌属模式菌株进行脂肪酸组成分析,并与16S rRNA系统进化分析进行比较,旨在为揭示基于脂肪酸生物标记的芽胞杆菌鉴定的准确性,基于脂肪酸生物标记的芽胞杆菌系统发育与芽胞杆菌生物学演化的关系,与16S rRNA芽胞杆菌系统发育的差异提供参考。
1 材料与方法
1.1 材料
1.1.1 供试菌株 所有供试菌株来自于本实验室福建省农业科学院农业生物资源所农业生物研究中心,分别引自德国微生物菌种保藏中心(DSMZ)、美国典型培养物保藏中心(ATCC)和瑞典Geborg大学菌物保藏中心(CCUG),详见表1。

表1 供试菌株信息
1.1.2 主要试剂和仪器 皂化试剂(试剂I):150mL去离子水和150 mL甲醇混匀合,加入45 g NaOH,同时搅拌至完全溶解;甲基化试剂(试剂II):325 mL 6 mol/L的盐酸加入到275 mL甲醇中,混合均匀;萃取试剂(试剂III):加200 mL甲基叔丁基醚到200 mL正己烷中,混合均匀;洗涤试剂(试剂IV):在900 mL去离子水中加入10.8 g NaOH,搅拌至完全溶解;饱和NaCl溶液:在100 mL去离子水中加入40 g NaCl。以上所有有机试剂均为色谱(HPLC)级,购于Sigma公司,无机试剂均为优级纯。安捷伦7890 N型气相色谱、Sherlock MIS、振荡器、水浴锅、10 mL带盖试管、玻璃量筒等。所有的玻璃器皿均须烘干后使用。
1.2 方法
1.2.1 菌株活化和保存 所有供试菌株均采用TSA(BD,US)培养基进行活化,28℃培养2 d。采用-80℃甘油冷冻法保存供试菌株,进一步试验备用。
1.2.2 芽胞杆菌脂肪酸的提取、检测及分析
1.2.2.1 菌体获取及脂肪酸的提取 参照文献[4]的方法进行。在TSB培养基上新鲜培养的待测菌株按四区划线接种至新鲜TSB培养基,28℃培养24 h。刮取20 mg菌体,置于试管,加入1 mL溶液I,100℃水浴30 min。冰浴中迅速冷却后加入2 mL溶液II,混匀后80℃水浴作用10 min。迅速冷却,加入1.25 mL溶液III。震荡10 min,吸弃下层溶液。然后中加入3 mL溶液IV及两滴饱和NaCl溶液,震荡5 min。静止,待溶液分层后,吸取上层液体于GC样品管中待测。
1.2.2.2 细菌脂肪酸成分检测 采用美国Agilent 7890 N型气相色谱系统,包括全自动进样装置、石英毛细管柱及氢火焰离子化检测器;通过细菌细胞脂肪酸成分进行细菌鉴定的分析软件采用Sherlock MIS6.0(Microbial Identification System)(美国MIDI公司产品)。在下述色谱条件下平行分析脂肪酸甲酯混合物标样和待检样本:二阶程序升高柱温,170℃起始,每分钟升温5℃,升至260℃,而后再每分钟升温40℃,升至310℃,维持90 s;汽化室温度250℃、检测器温度300℃;载气为氢气(2 mL/ min)、尾吹气为氮气(30 mL/min);柱前压68.95 Kpa;进样量1 μL,进样分流比100∶1。
1.2.2.3 统计分析 利用林营志等[9]编程软件PLFAEco处理后提取出脂肪酸数据,然后利用生物统计软件SPSS16.0进行统计分析。
1.2.3 基于16S rRNA基因序列的芽胞杆菌系统发育分析 芽胞杆菌模式菌株序列来自EzTaxon网站[10]。16S rRNA序列经过ClustalX2[11]程序多重比对,系统进化矩阵根据Jukes-Cantor模型[12]估计,利用Mega4.0软件[13]采用邻接法(Neighbour-Joining)[14]进行聚类分析构建系统进化树。同时采用1 000次自展值(Bootstrap value)分析来评估系统进化树拓扑结构的稳定性[15]。
2 结果
2.1 芽胞杆菌的脂肪酸成分分析
对25种芽胞杆菌进行了脂肪酸成分的测定,检测出22种已知脂肪酸和4种Summed Feature型,每种芽胞杆菌的具体脂肪酸成分见表 2。
表2显示,芽胞杆菌的脂肪酸主要以支链脂肪酸为主,少数种类含有不饱和脂肪酸,主要类型即脂肪酸含量大于10%的为15:0 iso、15:0 anteiso、17:0 iso、14:0 iso、16:0、16:0 iso和 17:0 anteiso。不同芽胞杆菌种类的脂肪酸成分和含量不同,如蜡状芽胞杆菌类群主要脂肪酸类型为15:0 iso、17:0 iso、16:0、13:0 iso和Summed Feature 3(16:1 ω6c and/or 16:1 ω7c),其次为15:0 anteiso、17:1 iso ω5c、14:0 iso、17:0 anteiso和13:0 anteiso。球形芽胞杆菌类群B. fusiformis、B. odysseyi、B. sphaericus主要脂肪酸为15:0 iso、15:0 anteiso和16:0 iso,其次为17:0 iso、17:0 anteiso、16:1 ω7c alcohol、14:0 iso和16:0。枯草芽胞杆菌类群B. vallismortis、B. atrophaeus和B. mojavensis脂肪酸主要为15:0 iso、15:0 anteiso和17:0 anteiso,其次为17:0 iso、16:0 iso、16:0和14:0 iso。酸性芽胞杆菌B. acidicola和B. acidiproducens主要脂肪酸为15:0 anteiso和17:0 anteiso。嗜碱芽胞杆菌类群B. agaradhaerens、B. alacalphilus、B. alkalitelluris和B. fastidiosus脂肪酸主要为16:0、15:0 iso、15:0 anteiso和17:0 iso。短小芽胞杆菌类群B. altitudinis、B. pumilus和B. safensis脂肪酸主要为15:0 iso和15:0 anteiso, 其 次 为17:0 anteiso、17:0 iso、16:0 iso、16:0和14:0 iso。简单芽胞杆菌类群B. koreensis、B. aryabhattai、B. megaterium、B. muralis、B. simplex和B. novalis的主要脂肪酸为15:0 iso和15:0 anteiso,其次为17:0 anteiso、17:0 iso、16:0 iso、16:0、14:0和14:0 iso。

表2 供试菌株的脂肪酸含量
2.2 基于脂肪酸生物标记的芽胞杆菌系统发育分析
利用生物统计软件SPSS16.0采用欧式距离对25种芽胞杆菌进行脂肪酸聚类分析,可以分为两大类,具体见图 1。第一类包含13种菌,进一步可分为4个小分支:B. safensis、B. pumilus和B. altitudinis聚为一个分支,B. aryabhattai、B. novalis和B. koreensis聚为一个分支,B. odysseyi、B. sphaericus和B. fusiformis聚为一个分支,B. mycoides、B. thuringiensis、B. cereus和B. pseudomycoides聚为一个分支。第二类包含12种菌,可分为两个小分支:B. alcalophilus、B. fastidiosus、B. alkalitelluris、B. vallismortis和B. agaradhaerens聚为一个分支,B. acidicola和B. acidiproducens、B. atrophaeus和B. mojavensis、B. megaterium、B. simplex和B. muralis聚为一个分支。

图1 基于脂肪酸生物标记的芽胞杆菌聚类分析
2.3 基于16S rRNA基因芽胞杆菌系统发育分析
25种芽胞杆菌采用ClustalX 2软件进行多序列分析,并通过Mega4软件构建系统进化树(图2)。结果显示,芽胞杆菌16S rRNA分类将芽胞杆菌分为两大类,第一类包含4个小分支,分别为:B. fastidiosus、B. alkalitelluris、B. megaterium、B. aryabhattai和B. koreensis聚为一个分支,B. novalis、B. simplex和B. muralis聚为一个分支,B. odysseyi、B. sphaericus和B. fusiformis聚为一个分支,B. mycoides、B. thuringiensis、B. cereus和B. pseudomycoides聚为一个分支。第二类包含3个小分支,分别为:B. acidicola和B. acidiproducens聚为一个分支,B. alcalophilus和B. agaradhaerens聚为一个分支,B. safensis、B. pumilus和 B. altitudinis,B. atrophaeus、B. vallismortis和 B. mojavensis聚为一个分支。
2.4 芽胞杆菌脂肪酸与16S rRNA系统发育分析比较
基于脂肪酸生物标记的芽胞杆菌聚类与16S rRNA聚类分析相比有一些差异,表 3显示,脂肪酸聚类结果与16S rRNA不同的是B. novalis和B. megaterium在聚类树中的位置互换,B. fastidiosus、B. alkalitelluris、B. alcalophilus和B. agaradhaerens在脂肪酸聚类分析中仅聚为一个分支,而非16S rRNA进化树中的两个独立分支,B. vallismortis与后4种芽胞杆菌聚在一起。脂肪酸分类主要依据是生物学特性,而16S rRNA分类则主要依据DNA碱基相似性。
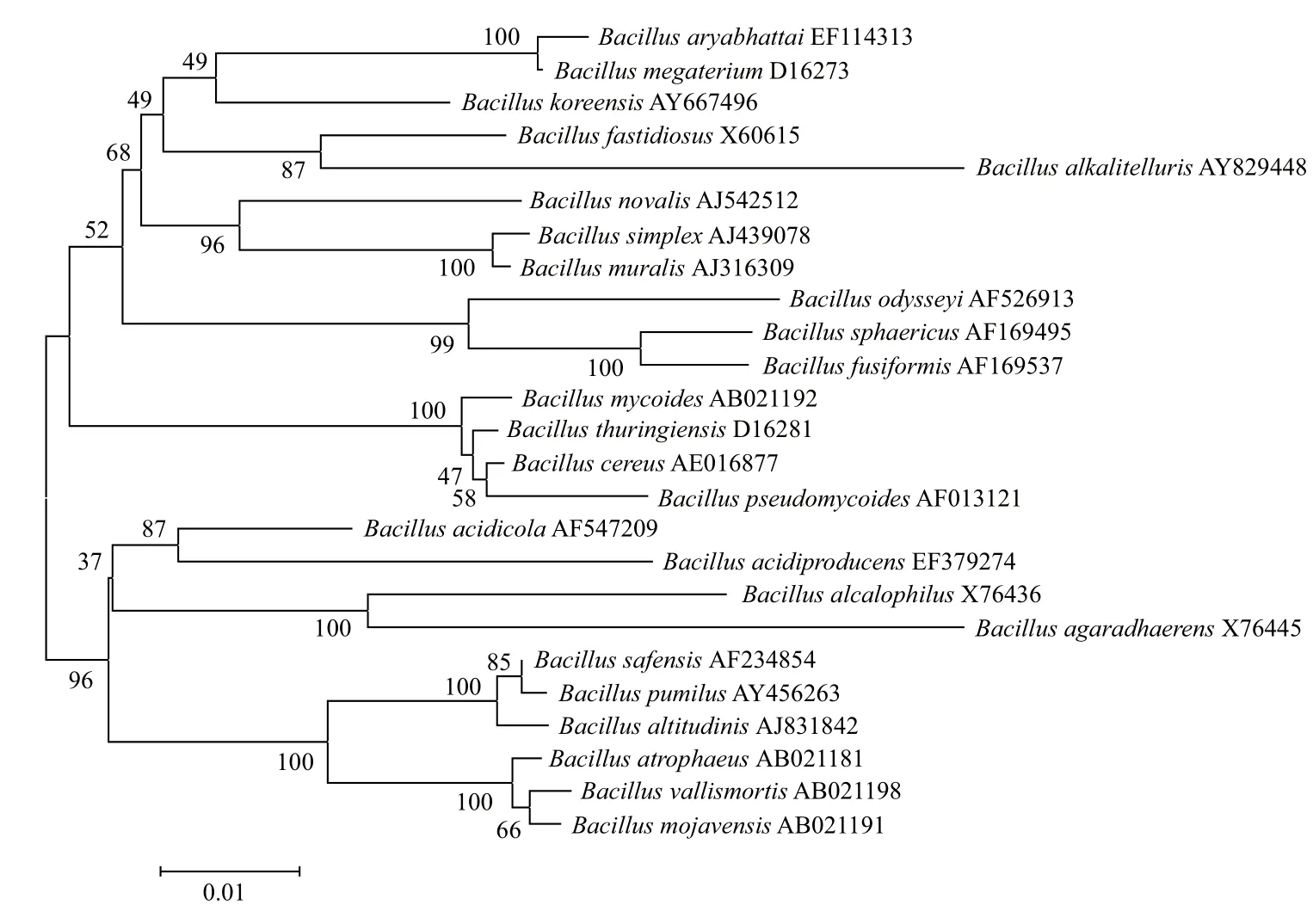
图2 基于16S rRNA芽胞杆菌系统进化分析
3 讨论
本研究针对25个芽胞杆菌种类进行了基于16S rRNA基因和脂肪酸生物标记系统发育分析的比较,结果表明,脂肪酸生物标记可以如16S rRNA基因一样为研究近亲缘物种之间的进化关系提供有用的信息,而且在有些种类的分群上脂肪酸更具有优势。随着数据库中芽胞杆菌脂肪酸数据的增加,可以进行更深入的研究。尽管脂肪酸提取分析需要较严格的操作标准,同时需要实验室具备专门的仪器,这些局限性随着科学技术和经济的发展都已逐步得到改善。脂肪酸鉴定已发展一种相对成熟的鉴定方法,在许多菌株中都曾有过应用。Abel 等[5]早在1963年就指出脂肪酸可用于细菌鉴定,细胞脂肪酸分析(Microbial identification system,MIDI)鉴定系统的应用使微生物脂肪酸分析标准化、自动化,操作简单,检测结果迅速而准确,且费用低廉,因此近些年被广泛地应用于细菌的分类鉴定中[16]。吴愉萍等[17]也将Sherlock MIS系统应用于土壤细菌鉴定的研究,结果表明该系统可以将分离菌株准确地鉴定到种,甚至可以进行种下分化鉴定分析。Whittaker等[18]研究证明脂肪酸分析可以快速灵敏地鉴定Francisella tularensis。刘波[19]出版的《微生物脂肪酸生态学》中,以本实验室分离芽胞杆菌为例,比较了脂肪酸鉴定与16S rRNA分子鉴定,结果表明,98%的芽胞杆菌种类用脂肪酸鉴定结果与16S rRNA分子鉴定结果相同,说明脂肪酸组成分析可快速而准确地对芽胞杆菌类群的菌株进行初步的鉴定。黄朱梁[20]用Sherlock微生物自动鉴定系统鉴定了从贻贝中分离的蜡样芽胞杆菌,并用生理生化鉴定和PCR鉴定验证了该方法的准确性。同时,通过对芽胞杆菌模式菌株的脂肪酸分析,我们首次报道了芽胞杆菌脂肪酸聚类分析与16S rRNA聚类结果的比较分析。
另外,由于16S rRNA基因高度保守,亲缘关系在种以上水平的菌株具有很高的分辨率,但对亲缘关系比较近的种分辨率不高。一般来讲,菌株之间16S rRNA基因序列相似性>97%,可能属于同一种;但16S rRNA基因序列相似性在99%以上,仍可能属于不同的种,需要DNA-DNA杂交等试验来进一步确定。基于16S rRNA的芽胞杆菌系统发育,唯一依据就是基因序列相似性,这一结果无法完美地体现芽胞杆菌生物学特性与系统发育的关系,如喜酸环境生存的芽胞杆菌B. acidicola、B. acidiproducens、B. atrophaeus和B. mojavensis等,喜碱环境生存的芽胞杆菌B. alcalophilus、B. fastidiosus、B. alkalitelluris和B. agaradhaerens等,不能聚为同一个分支。
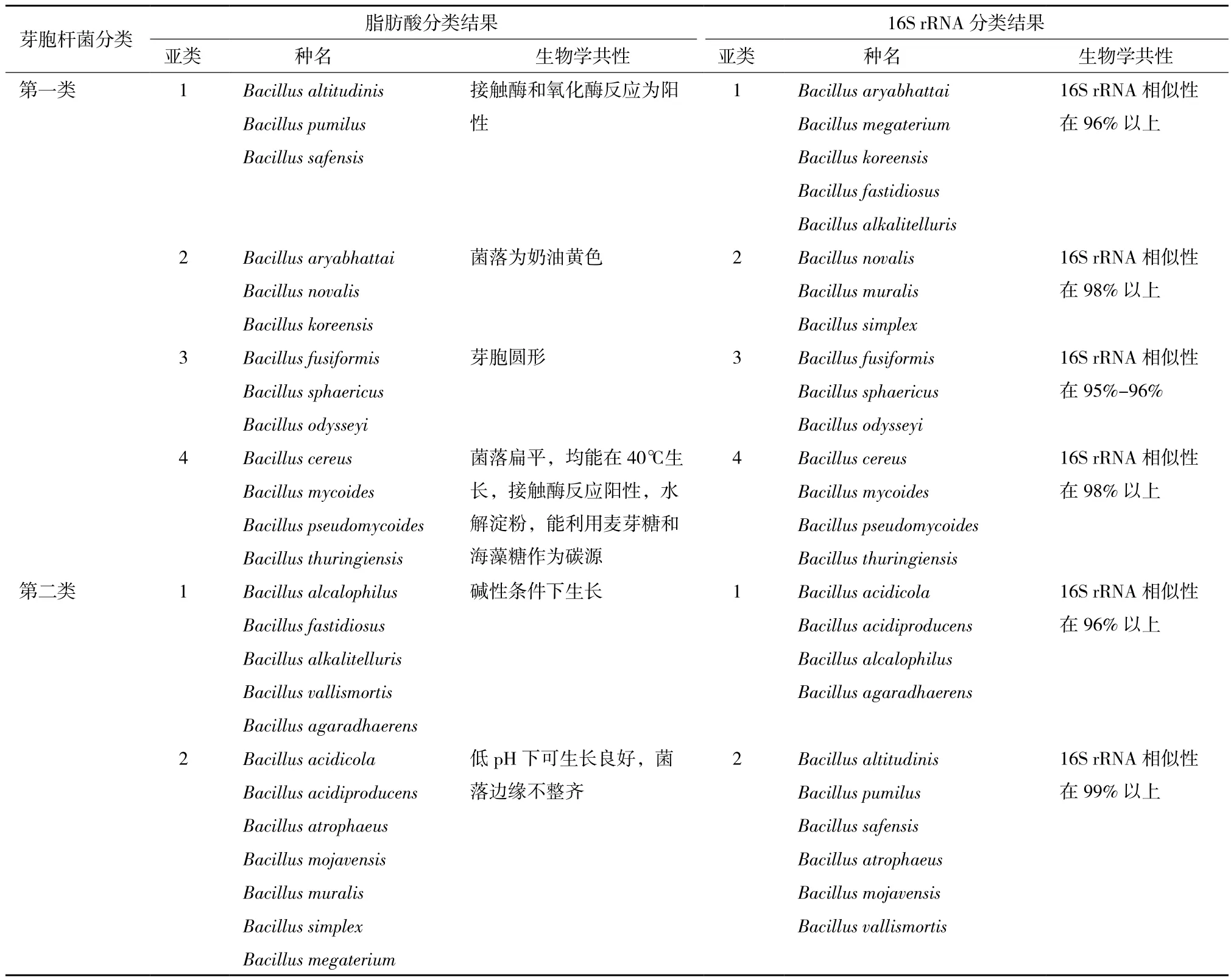
表3 芽胞杆菌脂肪酸和16S rRNA分类结果比较
本研究选取了25种芽胞杆菌模式菌株作为研究对象,比较分析了芽胞杆菌脂肪酸聚类和16S rRNA聚类分析的差异。研究发现利用脂肪酸组成分析也可以成功将供试菌株完全准确地区分开,这一点与16S rRNA的分类结果一致。我们还发现在芽胞杆菌“种”的分类地位上,脂肪酸聚类结果比16S rRNA基因进化分析更具有优势,不仅可以根据进化关系确定种的分类地位,还根据生物学特性将相同的菌株聚在一起,如本研究的供试菌株4种芽胞杆菌均为嗜碱菌,在16S rRNA进化分析中聚类为两个独立分支,而脂肪酸则根据其相同的生物学特性将其聚为一个分支。
本研究仅以芽胞杆菌属的种类为研究对象进行分析,对脂肪酸生物标记在芽胞杆菌近缘属上的应用是否具有同样的效果需要进一步研究验证。
4 结论
本研究比较分析了25种芽胞杆菌模式菌株脂肪酸组分和16S rRNA基因系统进化,结果表明脂肪酸分析可将25种芽胞杆菌完全准确地分开,且将生物学特性相同的芽胞杆菌种类聚为一类,如碱性条件下生长良好的4种芽胞杆菌(B. agaradhaerens、B. alacalphilus、B. alkalitelluris和B. fastidiosus)聚为一类,芽胞杆菌为圆形的芽胞杆菌(B. fusiformis、B. odysseyi 和B. sphaericus)聚为一类。
[1] 刘波, 朱昌雄. 微生物发酵床零污染养猪技术的研究与应用[M]. 北京:中国农业科学技术出版社, 2009.
[2] 车建美, 郑雪芳, 刘波, 等. 短短芽胞杆菌FJAT-0809-GLX 菌剂的制备及其对枇杷保鲜效果的研究[J]. 保鲜与加工, 2011,11(5):6-9.
[3] 刘波,刘国红,林乃铨. 基于脂肪酸生物标记的芽胞杆菌属种类的系统发育研究[J]. 微生物学报, 2014, 54(2):139-158.
[4] 陶天申, 杨瑞馥, 东秀珠. 原核生物系统学[M]. 北京:化学工业出版社, 2007.
[5] Abel K, Deschmertzing H, Peterson JI. Classification of microorganisms by analysis of chemical composition. I. Feasibility of utilizing gas chromatography[J]. J Bacteriol, 1963, 85(5):1039-1044.
[6] Kaneda T. Iso- and anteiso-fatty acids in bacteria:biosynthesis,function, and taxonomic significance[J]. Microbiological Reviews,1991, 55(2):288-302.
[7] Kämpfer P. Limits and possibilities of total fatty acid analysis for classification and identification of Bacillus species[J]. Syst Appl Microbiol, 1994, 17:86-98.
[8] 张晓霞, 王直强, 李世贵, 等. 脂肪酸组分分析在不动杆菌鉴定中的应用[J]. 生物技术通报, 2009(6):150-153.
[9] 林营志, 刘波, 张秋芳, 等. 土壤微生物群落磷脂脂肪酸生物标记分析程序PLFAEco[J]. 中国农学通报, 2009, 25(14):286-290.
[10] Kim OS, Cho YJ, Lee K, et al. Introducing EzTaxon-e:a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species[J]. Int J Syst Evol Microbiol, 2012,62 :716-721.
[11] Thompson JD, Gibson TJ, Plewniak F, et al. The CLUSTAL_ X windows interface:flexible strategies for multiple sequence alignment aided by quality analysis tools[J]. Nucleic Acids Res,1997, 25:4876-4882.
[12] Jukes TH, Cantor CR. Evolution of protein molecules[M]// Munro HN. Mammalian Protein Metabolism. New York:Academic Press, 1969:21-132.
[13] Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method[J]. Proceedings of the National Academy of Sciences(USA), 2004,101:11030-11035.
[14] Saitou N, Nei M. The neighbor-joining method:A new method for reconstructing phylogenetic trees[J]. Mol Biol Evol, 1987, 4:406-425.
[15] Felsenstein J. Confidence limits on phylogenies:an approach using the bootstrap[J]. Evolution, 1985, 40:783-791.
[16] Sasser M. Identification of bacteria by gas chromatography of cellular fatty acids[J]. USFCC News, 1990, 20:16.
[17] Whittaker P, Day JB, Curtis SK, et al. Evaluating the use of fatty acid profiles to identify Francisella tularensis[J]. J Microbiol Meth, 2007, 90(2):465-469.
[18] 吴愉萍, 徐建明, 汪海珍, 等. Sherlock MIS系统应用于土壤细菌鉴定的研究[J]. 土壤学报, 2006, 43(4):642-648.
[19] 刘波. 微生物脂肪酸生态学[M]. 北京:中国农业科学出版社,2011:1-798.
[20] 黄朱梁, 裘迪红. MIDI Sherlock微生物自动鉴定系统鉴定方法的建立[J]. 宁波大学学报:理工版, 2011, 24(2):8-13.
(责任编辑 马鑫)
The Comparsion of Bacillus Species Classification Based on Fatty Acid and 16S rRNA Gene
Liu Guohong Liu Bo Lin Yingzhi Tang Jianyang
(Agricultural Bio-resource Institute,Fujian Academy of Agricultural Sciences,Fuzhou 350003)
In order to explore the application of fatty acid composition in taxonomy of Bacillus species, the fatty acid constitutions of 25 strains were detected by Microbial Identification System(MIDI). The clusters of fatty acid profiles and 16S rRNA gene sequences were analyzed by SPPS16.0 and Mega4, respectively. The results showed that phylogeny analysis based on the fatty acid biomarkers could not only fully reflect the relationships among the Bacillus species, but also group the Bacillus species according to the biological characteristics. However,16S rRNA phylogeny only perfectly showed the relationships among the species. For example, four species growing well under the alkaline conditions and three species round spore-forming were clustered together, respectively. Result showed that Bacillus species can be clustered together not only according to the related ship, but also classified by the biological characteristics.
Bacillus;fatty acid profile;16S rRNA;phylogeny
10.13560/j.cnki.biotech.bull.1985.2015.04.021
2014-08-05
国家自然科学基金项目(31370059),“973”前期研究专项(2011CB111607),农业部“948计划”(2011-G25)
刘国红,博士,研究方向:芽胞杆菌资源分类及功能研究;E-mail:Liuguohong624@sina.com
刘波,博士,研究员,研究方向:微生物生物技术与农业生物药物;E-mail:fzliubo@163.com
