两种膨胀单体的合成及其对牙科修复树脂的改性
2014-06-11张露邱婷邢晓东
张露,邱婷,邢晓东
(南京理工大学化工学院,江苏 南京 210094)
自1962年美国学者Bowen发明牙科复合树脂以来,由于其操作简便,色泽美观,并且弥补了以前树脂在硬度和耐磨性方面的不足,其应用发展一直备受关注[1-2]。商用牙科复合树脂单体主要基于甲基丙烯酸酯类单体Bis-GMA及其衍生物[3]。然而,随着临床的广泛应用,树脂修复材料的一些缺点也逐渐暴露,其中比较重要的是Bis-GMA等单体在聚合过程伴随着较大的体积收缩[4],易导致树脂修复体与牙体粘接面产生微渗漏而造成继发龋[5-6],严重者甚至出现粘接断裂,造成牙科修复治疗失败[5]。多年来,致力于低收缩树脂的研究大多都集中在对传统丙烯酸酯类单体的改性上[7]。
膨胀单体是一类在聚合过程中会产生体积膨胀的环状化合物,自1972年Bailey[8]发现膨胀单体以来,迄今已有多种膨胀单体被合成出来。基于相同的聚合机理以及良好的相容性,膨胀单体对环氧树脂的收缩改性很早就取得成功[9],近年来其在改性牙科修复树脂方面的研究也有少量报道[10-12]。
基于膨胀单体改性环氧树脂收缩的成功先例以及混杂体系在不同聚合反应机制下互相促进的效果[13],本研究合成了两种不同侧链结构的螺环原碳酸酯类膨胀单体,探讨了其改性环氧树脂单体与牙科用丙烯酸类树脂单体组成的混杂反应体系的聚合情况,以及对聚合反应产生的体积收缩的改善效果。
1 实验部分
1.1 主要仪器及试剂
三羟甲基丙烷单烯丙基醚(TMPME),纯度98%,天津西恩思生化试剂有限公司;原碳酸四乙酯(TEOC),纯度99%,成都格雷西亚化学技术有限公司;对甲苯磺酸(PTSA),分析纯,上海凌峰化学试剂有限公司;三羟甲基丙烷(TMP),分析纯,阿拉丁试剂有限公司;二正丁基氧化锡(DBTO),纯度98%,上海岚克生物医药有限公司;CS2,分析纯,阿拉丁试剂有限公司;3,4-环氧环己基甲基 3,4-环氧环己基甲酸酯(EE),纯度>98%,阿拉丁试剂有限公司;甲基丙烯酸树脂,双酚A甲基丙烯酸缩水甘油酯(Bis-GMA),纯度>99%,Sigma-Aldrich公司;三乙二醇二甲基丙烯酸酯(TG),纯度>98%,日本东京化成工业株式会社;二苯基碘鎓六氟磷酸盐(PI),纯度>99%,天津西恩思生化试剂有限公司;樟脑醌(CQ),纯度>98%,阿拉丁试剂有限公司;甲基丙烯酸二甲氨基乙酯(DMAEMA),分析纯,阿拉丁试剂有限公司,减压蒸馏纯化后使用;混杂引发剂配比为 PI∶CQ∶DMAEMA=3∶1∶0.1(质量比,下同)。
Bruker Avance III 500MHz核磁共振仪;Thermo Fisher Nicolet IS-10型傅里叶变换红外光谱仪;X-5显微熔点测定仪;LED可见光固化灯(波长465~480 nm);华银HV-10B显微硬度计。
1.2 膨胀单体的合成
1.2.1 3,9-二乙基-3,9-二羟甲基-1,5,7,11-四氧螺杂[5,5]十一烷(DHOM)的合成
[14]的方法并加以改进,称取6.709 g TMP和12.45 g DBTO,80 mL甲苯为溶剂搅拌加热至120 ℃,回流分水16 h以上降至室温,拆除分水器,换上干燥回流冷凝管,从恒压滴液漏斗缓慢滴加CS2约5 mL,滴加完毕后重新升温至100 ℃搅拌回流 12 h后停止。产物减压浓缩得黏稠液体,用正己烷洗涤2~3次,以甲苯加热溶解,冷却重结晶,抽滤并烘干后得产物 DHOM 5.82 g,产率为84.3%。
1.2.2 3,9-二乙基-3,9-丙烯氧甲基-1,5,7,11-四氧螺杂[5,5]十一烷(BAOM)的合成
参考文献[15]的方法并加以改进,称取5.236 g TMPME,加入80 mL甲苯为溶剂,磁力搅拌加热至111 ℃,回流分水2 h后降至室温,加入0.06 g PTSA,滴加3 mL TEOC,重新升温至111 ℃以上搅拌回流分出副产物乙醇,反应24 h后降至室温,滴加1 mL三乙胺室温搅拌1 h。产物减压浓缩,经柱色谱(乙酸乙酯∶石油醚=1∶30)分离纯化得BAOM 4.88 g,产率为91.1%。
1.3 膨胀单体/环氧树脂对Bis-GMA树脂体系的改性
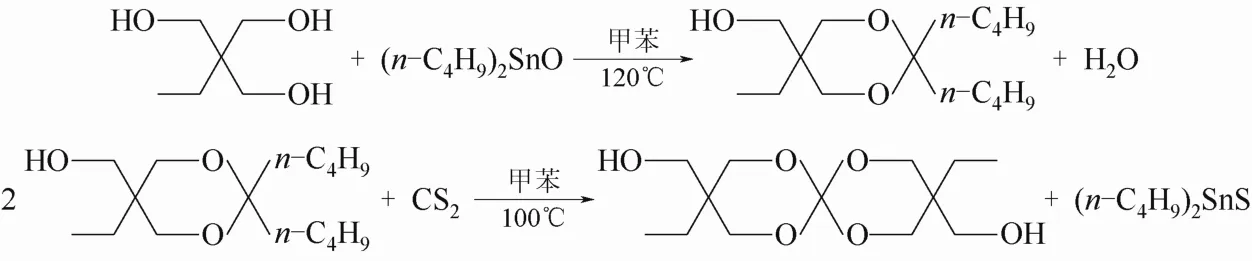
图1 DHOM 的合成路线
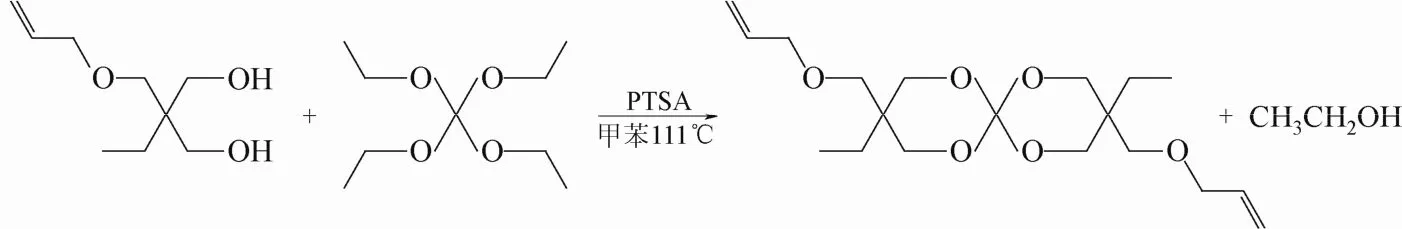
图2 BAOM 的合成路线
分别将两种膨胀单体DHOM及BAOM以不同质量比与EE混合,添加质量分数5%的引发剂,搅拌均匀后固化照射80 s,测定EE在不同含量膨胀单体改性后聚合的体积收缩率[16],确定两种单体改性EE的最佳比例。
以上述确定的DHOM和BAOM与EE的最佳比例分别配制改性环氧树脂混合液 DHOM-EE和BAOM-EE。由于Bis-GMA黏度较大,临床常以二缩三乙二醇双甲基丙烯酸酯(TG)稀释,二者质量比 Bis-GMA∶TG=3∶1。将混合液 DHOM-EE和BAOM-EE分别以不同质量比与 Bis-GMA/TG混合,添加5%的引发剂,搅拌均匀固化照射80 s,测定固化前后的体积收缩率,并用FT-IR表征固化前后体系结构变化与聚合双键转化率[17]。
将混合液BAOM-EE以不同质量比与未稀释的Bis-GMA混合,添加5%的引发剂,搅拌均匀固化照射80 s,测定固化前后的体积收缩率,用FT-IR表征固化前后体系结构变化与聚合双键转化率,并测定改性前后树脂的硬度变化。
2 结果与讨论
2.1 膨胀单体的结构与表征
2.1.1 DHOM的结构与表征
DHOM 为白色粉末,熔程为 59.2~60.4 ℃。图3为DHOM的FT-IR谱图。3567 cm−1处尖峰为羟基O—H伸缩振动,3400 cm−1处宽峰为分子间氢键O—H伸缩振动,表明结构中存在羟基;1011~1264 cm−1间的峰为C—O伸缩振动峰,1220 cm−1附近是具有明显的螺环基团的特征吸收峰,表明分子中螺环结构的存在。
图4为DHOM的1H NMR谱图,0.81~0.86为甲基质子峰(6H),1.34~1.41处是乙基上的亚甲基质子峰(4H),2.7~2.9为羟基质子峰(2H),3.6~3.8为与氧相连的亚甲基质子峰(12H)。

图3 DHOM的FT-IR谱图

图4 DHOM的1H NMR谱图
2.1.2 BAOM的结构与表征
BAOM为低黏度无色液体。图5为BAOM的FT-IR谱图。2966~2881cm−1处为饱和烃 C—H伸缩振动,1363~1455cm−1处为C—H弯曲振动,表明—CH3,—CH2—的存在;1228 cm−1处为螺环特征吸收峰;1647 cm−1为C=C的伸缩振动,表明烯键的存在;1068 cm−1附近出现侧链状醚键,证实了该单体的分子结构。

图5 BAOM的FT-IR谱图

图6 BAOM的1H NMR谱图
图6为BAOM的1H NMR谱图。0.81~0.85处为侧链乙基上的甲基质子峰(6H),1.36~1.42处为乙基上的亚甲基质子峰(4H),3.48为链上与氧相连的亚甲基质子峰(4H),3.68~3.85处为螺环上与氧相连的亚甲基质子峰(8H),3.97~3.99处是与氧和烯键相连的亚甲基质子峰(4H),5.14~5.28处为乙烯基上的甲基质子峰(4H),5.84~5.93处为乙烯基上的亚甲基质子峰(2H)。
2.2 膨胀单体改性环氧树脂体系的确立
图7为不同质量的两种膨胀单体与环氧单体EE的混合后,聚合前后的体积收缩率变化。
由于 DHOM 为固体粉末,随其添加量增加,混合体系黏度逐渐增大,超过60%呈现凝固态。如图7中曲线a所示,随DHOM添加量从0升至60%,体系的聚合收缩率从 4.77%降至 2.21%,但树脂体系黏度过大会给其临床使用带来极大的不便,因此综合考虑混合体系的黏度和体积收缩值,DHOM改性环氧树脂的最佳比例定为DHOM∶EE=1∶1。
BAOM为低黏度液体,且与EE互溶性良好。如图7中曲线b所示,随BAOM添加量从0升至50%时,体系聚合前后的收缩率从 4.77%降至2.54%,当其添加到60%后,收缩率反而有所回升,原因可能是 BAOM 添加量过多限制了自身的开环反应,导致聚合收缩反而增大。因此BAOM改性环氧树脂的最佳比例为BAOM∶EE=1∶1。
2.3 改性Bis-GMA牙科修复树脂体系的确定
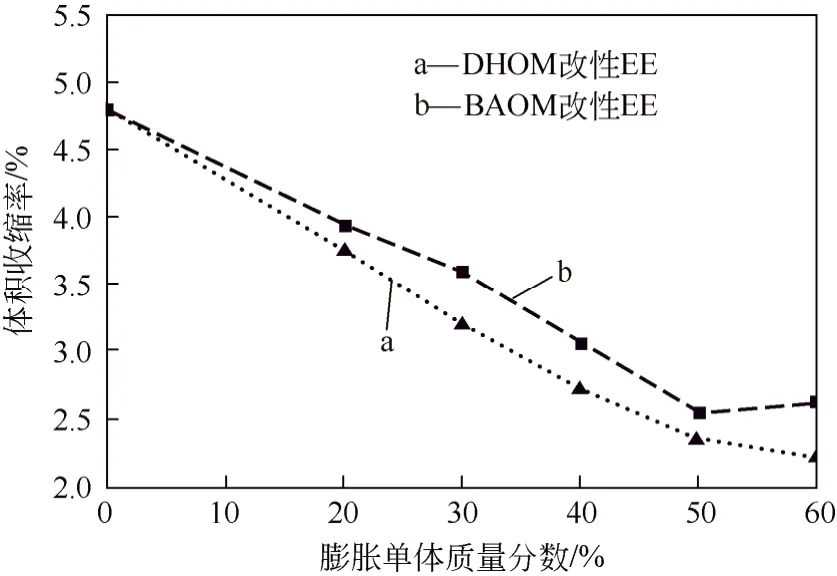
图7 膨胀单体改性环氧树脂体系体积收缩变化
用以上述两种膨胀单体改性EE的最佳比例配置改性混合液DHOM-EE和BAOM-EE,图8显示了DHOM-EE和BAOM-EE改性牙科修复树脂体系聚合后的体积收缩率变化。如图8中曲线a、曲线b所示,未添加改性的Bis-GMA/TG收缩率6.00%。随DHOM-EE和BAOM-EE添加量从0增加到60%,Bis-GMA/TG收缩率变化趋势相同,均为添加初始显著降低,添加量增大到一定程度后趋于不变,DHOM-EE添加到 60%后收缩率可降至 2.97%。BAOM-EE添加到 60%后收缩率可降至 3.06%。DHOM-EE对稀释后的Bis-GMA树脂体系改性效果整体略优于BAOM-EE。

图8 膨胀单体改性Bis-GMA树脂体系体积收缩变化
由于BAOM-EE为低黏度液体,添加后可以显著降低体系浓度,因此本研究将其用于直接改性未经稀释的 Bis-GMA,如图 8中曲线 c所示,随BAOM-EE添加量从0增加到60%,收缩率从5.68%降至 2.72%,不仅进一步改善了体系的收缩,而且替代了TG作为稀释剂,达到了稀释和收缩改性的双重效果。
2.4 膨胀单体改性Bis-GMA树脂体系的FT-IR
2.4.1 改性体系固化前后的FT-IR
本实验所测的树脂体系为甲基丙烯酸树脂和环氧树脂以及膨胀单体组成的共混体系。其中甲基丙烯酸树脂的固化方式为自由基引发的加成聚合[18]。环氧树脂与膨胀单体属于阳离子开环聚合[19]。

图9 50%BAOM-EE改性Bis-GMA固化前后FT-IR图
图9、图10分别为两种膨胀单体改性前后树脂体系的FT-IR谱图。两图均显示改性树脂固化后,1638 cm−1附近碳碳双键的特征吸收峰明显减弱,表明自由基聚合反应的发生;螺环开环所生成的碳酸酯羰基在1722 cm−1处吸收峰增强,1065 cm−1处链状醚键有所上升,表明膨胀单体和环氧树脂阳离子开环反应的进行,FT-IR谱图表明改性树脂体系固化为两种聚合机理共存的混杂光固化。

图10 50%DHOM-EE改性Bis-GMA/TG固化前后FT-IR图
2.4.2 改性前后树脂体系的双键转化率
以不受固化反应影响的苯环(1610 cm−1)为参照,由1638 cm−1的“C=C”双键峰面积的变化计算出聚合体系的双键转化率。分别测定改性修复树脂体系固化照前、后的红外光谱,将所得数据进行分峰拟合处理,计算双键转化率结果如表1、表2所示。
由表1、表2可知,随着两种膨胀单体和环氧单体EE的引入,树脂体系的双键转化率明显升高。表明了阳离子开环聚合体系的引入不但没有阻碍Bis-GMA自由基聚合体系的反应,反而使双键转化率明显上升,自由基聚合度明显增大。

表1 膨胀单体-环氧改性Bis-GMA/TG体系的双键转化率

表2 BAOM-EE改性Bis-GMA体系的双键转化率
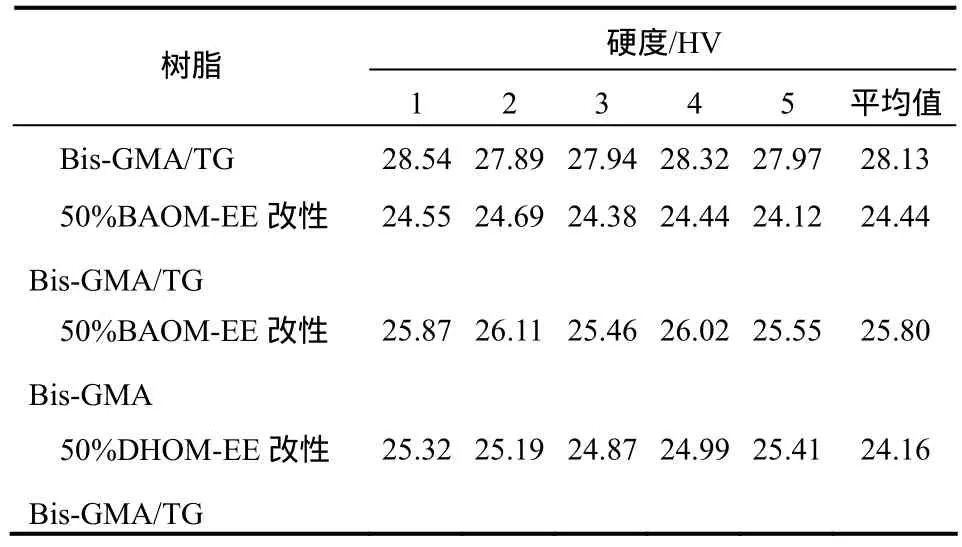
表3 改性前后树脂的硬度值
2.5 改性树脂体系的硬度
以聚四氟乙烯模具制备圆柱形试件(l = 8 mm,d = 2 mm),置于显微硬度仪上测试维氏硬度值,载荷6 kg,加载停留10 s。
表3列出了改性前后体系聚合后的硬度值。可知经膨胀单体和环氧树脂改性后,树脂的硬度较改性前均略有下降,原因可能是双酚A甲基丙烯酸缩水甘油酯(Bis-GMA)结构中含有刚性苯环结构,而改性后苯环的含量有所下降。经过 50%BAOMEE和50%DHOM-EE改性后的树脂硬度差别不大,表明两种单体对于改性体系的影响类似。而50%BAOM-EE改性 Bis-GMA的硬度高于改性Bis-GMA/TG,原因可能是因为不再添加稀释剂三乙二醇二甲基丙烯酸酯(TG)后,体系中苯环结构比例相对增多。
3 结 论
合成了两种不同结构与性状的膨胀单体BAOM 和 DHOM,并将其与环氧树脂单体混合用于传统牙科树脂体系的改性,研究结果表明改性树脂的聚合体积收缩值均明显降低,DHOM较BAOM改性效果更佳,最低可由原来的6%降至2.97%。但BAOM因黏度较低,可替代稀释剂TG,因此可进一步降低收缩至2.73%。FT-IR谱图表明树脂的自由基聚合程度较未改性的甲基丙烯酸树脂有明显上升。改性后树脂的硬度较之前略有下降。本研究表明,两种膨胀单体改性传统牙科树脂后不但降低了树脂的聚合收缩,还可增强树脂体系的聚合程度,而此改性对树脂的硬度影响也不大,有望用于开发新型的低收缩牙科修复树脂材料。
参 考 文 献
[1]Thompson V P,Williams E F,Bailey W J. Dental resins with reduced shrinkage during hardening[J]. Journal of Dental Research,1979,58(5):1522-1532.
[2]Scherrer S S,Cesar P F,Swain M V. Direct comparison of the bond strength results of the different test methods:A critical literature review[J]. Dental Materials,2010,26(2):e78-e93.
[3]Ferracane J L. Using posterior composites appropriately[J]. The Journal of the American Dental Association,1992,123(7):53-58.
[4]张盛炎,杨军英,王海燕,等. 三种后牙树脂聚合体积收缩及显微硬度的性能比较[J]. 中国组织工程研究与临床康复 ISTIC,2007,11(40):8043-8046.
[5]Bouillaguet S,Wataha J C. Future directions in bonding resins to the dentine-pulp complex[J]. Journal of Oral Rehabilitation,2004,31(4):385-392.
[6]刘文英,张文云. 复合树脂修复中继发龋的影响因素[J]. 西南国防医药,2011,21(2):217-219.
[7]Braga R R,Ferracane J L. Alternatives in polymerization contraction stress management[J]. Critical Reviews in Oral Biology & Medicine,2004,15(3):176-184.
[8]Bailey W J. Synthesis of monomers that expand on polymerization[J].Journal of Elastomers and Plastics,1973,5(3):142-152.
[9]周志强,何平笙,潘才元. 固化时不收缩胶粘剂的研制与应用[J].工程塑料应用,1989(3):31-34.
[10]Ortiz R A,Medina L A R,Duarte M L B,et al. Synthesis of glycerol-derived diallylspiroorthocarbonates and the study of their antishrinking properties in acrylic dental resins[J]. Journal of Materials Science:Materials in Medicine,2013,24(8):2077-2084.
[11]付静,贾芳,刘小青. 螺环原酸酯对牙科基体树脂的改性研究[J].临床口腔医学杂志,2010,26(2):80-83.
[12]Sun X,Li Y,Xiong J,et al. Shrinkage properties of a modified dental resin composites containing a novel spiro-orthocarbonate expanding monomer[J]. Materials Letters,2011,65(23):3586-3589.
[13]Gómez M L, Previtali C M , Montejano H A. Two-and three-component visible light photoinitiating systems for radical polymerization based on onium salts:An overview of mechanistic and laser flash photolysis studies[J]. International Journal of Photoenergy,2012,2012:1-9.
[14]白如科,王长松. 含螺环原碳酸酯齐聚物的合成表征及与环氧树脂的固化反应[J]. 功能高分子学报,1995,8(3):321-327.
[15]Chappelow C C,Pinzino C S,Chen S S,et al. Photopolymerization of a novel tetraoxaspiroundecane and silicon containing oxiranes[J].Journal of Applied Polymer Science,2007,103(1):336-344.
[16]于智,智放,王长松,等. 一种新膨胀单体的合成及其预聚物与环氧树脂的共聚研究[J]. 沈阳化工学院学报,2003,17(2):126-128.
[17]Stansbury J W,Dickens S H. Determination of double bond conversion in dental resins by near infrared spectroscopy[J]. Dental Materials,2001,17(1):71-79.
[18]Peutzfeldt A. Resin composites in dentistry:The monomer systems[J].European Journal of Oral Sciences,1997,105(2):97-116.
[19]Miller M D,Holder A J,Chappelow C C,et al. A theoretical study of an expanding monomer and an oxirane Part 1:Expanding monomer reactions[J]. Journal of Molecular Structure:Theochem.,2005,756(1):185-194.
