间二甲苯分子结构和红外光谱的密度泛函理论计算的研究
2013-11-23程海涛吴佩远
程海涛,吴佩远,田 仲,王 蕾
(衡水学院 化工学院,河北 衡水 053000)
间二甲苯是重要的化工原料之一,可用于石化、医药、纺织、农药、精细化工等多种行业.主要用作化工产品的异构化原料、医药、合成农药中间体、作为溶剂调节成品油的组成、合成高分分子聚合物树脂、合成其它精细化工产品.间二甲苯的最大用途是直接合成间苯二甲酸(IPA),IPA进一步合成不饱和聚酯树脂,不饱和聚酯树脂广泛应用于建筑、交通和海洋等领域.由于国民经济、交通运输等方面的飞速发展,对于不饱和聚酯树脂的需求与日俱增,带动间二甲苯的生产迅猛发展.因此,对间二甲苯的分子结构和红外光谱进行深入细致的研究,得到稳定构象,对于生成间二甲苯以及合成其它中间体、衍生物有重要意义,为更精确地控制各种合成过程的变化提供重要理论依据,意义十分重大.
密度泛函数理论(density functional theory, DFT),在电子互换和有关分子能量方面,有系统全面的考虑,使计算结构更加接近实际.密度泛函数理论广泛用于分子结构和光谱性质的研究和探讨,是一条行之有效的途径[1-2].
本文利用密度泛函数理论(DFT)-B3LYP方法,在6-31 + G(d)基组水平上,计算了对二甲苯分子结构参数和红外光谱,期望为进一步研究有关间二甲苯合成反应的机理提供有参考价值的结构与光谱信息.
1 计算方法
根据参考文献[3]提供的间二甲苯的分子结构图,采用Gaussian03 view构图软件构件分子结构的模型,同时利用Gaussian03对结构进行优化,在优化的基础上,采用密度泛函理论(DFT)-B3LYP的方法,在6-31 + G(d)水平上对其进行振动频率计算,得到稳定的构象、振动频率和红外光谱强度,计算是在PC-intel(R)Core(TM)i3-2120 2.0G上完成的.
2 结果与讨论
经过上述方法计算,得到12种优化构型,各种构型的总能量如表1所示.
2.1 间二甲苯分子几何构型的优化
以密度泛函数理论(DFT)基础,利用 B3LYP/6-31+G(d)基组水平计算方法对间二甲苯分子构型进行优化处理,对12种优化构型进行分析,总能量为-310.895 598 3 J的构型,分子能量最小,构型最稳定(如图1).表2中列出了间二甲苯最稳定构象的结构参数,包括键长、键角和二面角.从Gaussian03计算得到的结构参数看出,苯环上的C-C单键比单独烷烃的C-C单键的键长(0.154 0 nm)短一些,和单独的双键(0.134 0 nm)相比,键长要长一些,即环上原子间的键长存在单双键之间平均化的趋势,从计算得到的二面角数值可以看出,二面角几乎均接近 0°和 180°,由此推断环上原子均以 SP2的杂化方式形成了平面的结构构型,剩余没有杂化的 P轨道相互之间平行连接形成了闭合的共轭大π键.
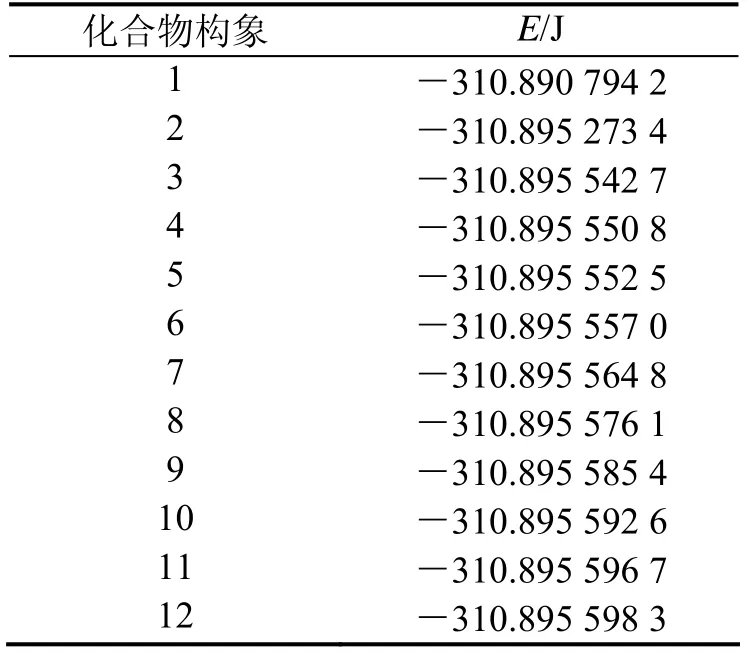
表1 间二甲苯不同构象的总能量

表2 间二甲苯最优结构在B3LYP/6-31+G(d)水平上的结构参数
2.2 间二甲苯的红外光谱
经过Gaussian03计算,结果得到间二甲苯的48种简正振动模式,没有任何虚频出现,由此证明了计算得到的最佳几何构型的位能最小.间二甲苯分子的简正振动频率处于400~3 300 cm-1范围之内,其中包含43个波数在400 cm-1之上的简正振动模式.利用(DFT)-B3LYP计算方法得出的间二甲苯分子IR光谱图如图2所呈现.表 3列出了利用(DFT)-B3LYP计算方法得到的振动光谱频率和强度.从计算得到的间二甲苯红外光谱图2,我们可以清晰地看到在400~3 300 cm-1之间有许多间二甲苯的IR特征吸收光谱区.在可视化软件的帮助下对于各频率的振动情况进行了细致深入的研究,对振动强度大的频率的归属逐一进行了解析:444 cm-1为C1—H7,C6—H10,C5—H9,C11—12,C3—H8,C15—H16,C15—17 的面内振动;703 cm-1为 C1—C2—C3—C4—C5—C6—C15—C11的面外振动以及苯环C—H的伸缩振动,甲基C—H的面内摆动;784 cm-1为苯环上的C-H伸缩振动和甲基(C15)的面内摆动和C11—H13的面外振动;1 508 cm-1为苯环及两个甲基的面内摆动;1 525 cm-1为甲基的面内摆动同时伴有C1—C2—C15,C4—C5—C11的变形振动;1 658 cm-1苯环的变形振动1 537 cm-1为苯环双键和芳基的C—H面内振动;3 086为甲基的C11—H14,C11—H12伸缩振动.利用B3LYP/6-31+G(d)计算方法得出的间二甲苯分子IR光谱于标准谱图的特征峰有很好的一致性.
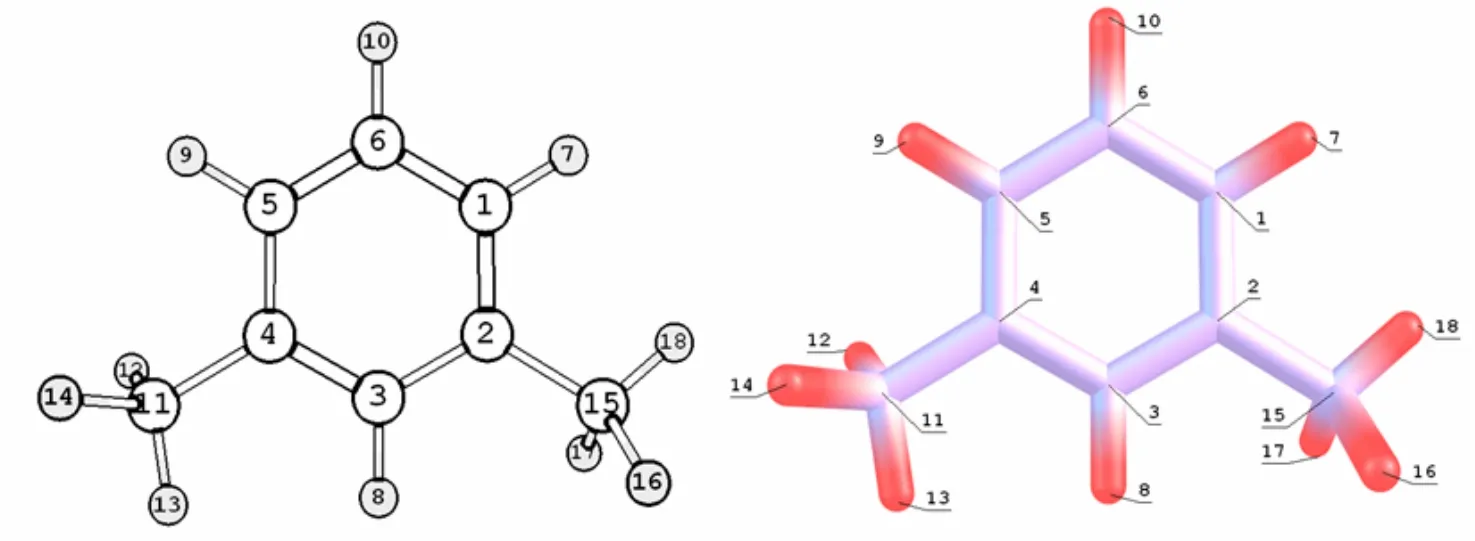
图1 间二甲苯分子结构和各原子标志

图2 间二甲苯在B3LYP/6-31+G(d)水平上计算得到的红外谱图

表3 间二甲苯的红外光谱频率和强度
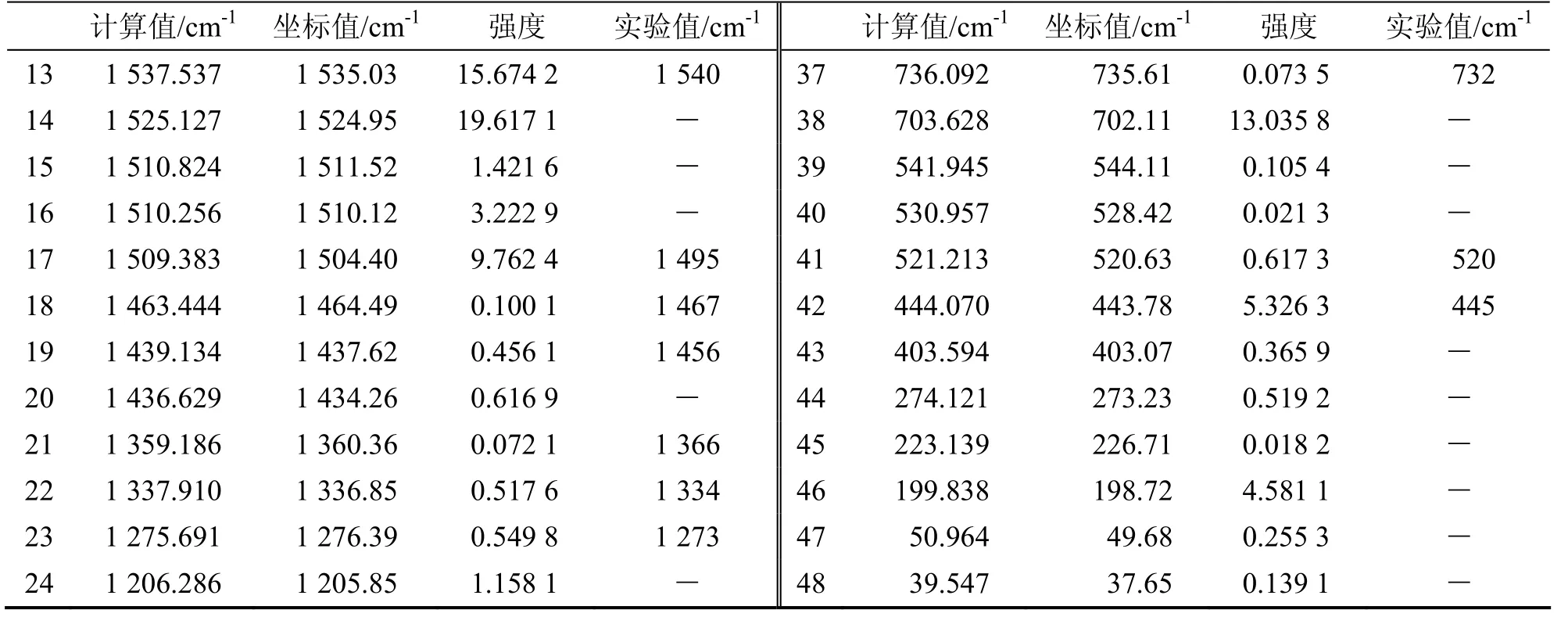
续表
3 结论
1) 采用密度泛函数理论(DFT)-B3LYP方法,在6-31+G(d)基组水平计算,得到了间二甲苯分子的最优结构构型.结果表明,间二甲苯分子的主要原子间的二面角基本上位0°或者180°.
2) 在同样的理论和基组水平下计算出了间二甲苯的 IR光谱,计算得出的振动频率与文献值一致程度很高,同时能够便捷的对振动频率的归属进行详细的解析.因此,密度泛函数理论(DFT)-B3LYP计算方法,可用于利用间二甲苯合成其它化合物机理模拟预测,为实际合成与化工生产以及构效关系深入研究提供有力的理论支撑.
