咪唑基离子液体与小分子相互作用的理论研究
2013-10-26吕仁庆
林 进,吕仁庆
咪唑基离子液体与小分子相互作用的理论研究
林 进1,*吕仁庆2
(1. 中国石油大学(华东)化学工程学院,山东,青岛 266580;2. 中国石油大学(华东)理学院,山东,青岛 266580)
采用密度泛函理论考察了1-乙基-3-甲基咪唑四氟合硼酸盐([EMIM][BF4])和2-甲基噻吩(2-MT)、正己烷(HEX)、异丁基硫醇(IBT)的相互作用。采用GGA/PW91和DNP基组优化了结构,并用NBO和AIM分析了[EMIM][BF4]和2-甲基噻吩(2-MT)、正己烷(HEX)、异丁基硫醇(IBT)的氢键相互作用。[EMIM][BF4]离子对最稳定的气相结构表明,[BF4]-阴离子的F原子和咪唑环上C2-H2的氢键作用在形成离子对中起重要作用。[BF4]-阴离子和[EMIM]+阳离子支链发生氢键作用。[BF4]-阴离子趋向于C2-H2形成氢键,这说明,2-甲基噻吩(2-MT)、正己烷(HEX)、异丁基硫醇(IBT)吸附在[EMIM][BF4]上没有改变离子液体离子对的主要作用。[EMIM][BF4]和2-甲基噻吩发生p···C-H和氢键作用,而[EMIM][BF4]和正己烷(HEX)、异丁基硫醇(IBT)主要发生氢键作用。相互作用能表明,2-甲基噻吩优先吸附在离子液体上。
离子液体;密度泛函理论;2-甲基噻吩;正己烷;异丁基硫醇
0 引言
在室温或室温附近温度下呈液态的由离子构成的物质,称为室温离子液体。2001年,Wasseerscheid等人[1]首先发现,离子液体可有效的脱除汽油中的含硫化合物。2003年,Lo等人[2]首先采用H2O2氧化和离子液体萃取的联合方法脱除汽柴油中的含硫化合物,发现比单一的离子液体萃取方法更有效。随后离子液体脱硫研究广泛展开[3-11]。最近,采用理论模拟方法考察了一些离子液体与含硫化合物的相互作用[12-16]。2002年,Zhang等人[17]考察了1-乙基-3-甲基咪唑四氟合硼酸盐离子液体的脱硫作用,研究表明,2-甲基噻吩的脱除效果远远比异丁基硫醇的脱除效果要好。因此,我们采用密度泛函理论方法考察了2-甲基噻吩、异丁基硫醇与1-乙基-3-甲基咪唑四氟合硼酸盐离子液体的相互作用。考虑到正己烷经常被用作溶剂,同时考察了正己烷与1-乙基-3-甲基咪唑四氟合硼酸盐离子液体的相互作用。着重考察了小分子2-甲基噻吩、异丁基硫醇、正己烷与1-乙基-3-甲基咪唑四氟合硼酸盐离子液体相互作用的电子性质和拓扑性质。
1 初始模型的设计
1-乙基-3-甲基咪唑阳离子([EMIM]+)、四氟合硼酸根离子([BF4]-),2-甲基噻吩(2-MT)、正己烷(HEX)、异丁基硫醇(IBT)如图1所示。首先考察[EMIM]+和[BF4]-的所有可能的相互作用,分别将[BF4]-阴离子置于C2-H2、C4-H4、C5-H5、咪唑环上方或下方、乙基或甲基的附近,形成初始的[EMIM][BF4]结构;其次,将2-甲基噻吩、正己烷、异丁基硫醇分别置于最稳定的[EMIM][BF4]的周围,形成[EMIM][BF4]-2-MT、[EMIM][BF4]-HEX、[EMIM][BF4]-IBT的初始结构;再次,分别将2-甲基噻吩、正己烷、异丁基硫醇置于[EMIM]+周围不同的位置,然后再将[BF4]-置于它们的周围,形成[EMIM][BF4]-2-MT、[EMIM][BF4]-HEX、[EMIM][BF4]-IBT的初始结构;最后,分别将2-甲基噻吩、正己烷、异丁基硫醇置于[BF4]-的不同位置,让后再将[EMIM]+置于它们的周围形成[EMIM][BF4]-2-MT、[EMIM][BF4]-HEX、[EMIM] [BF4]-IBT的初始结构。
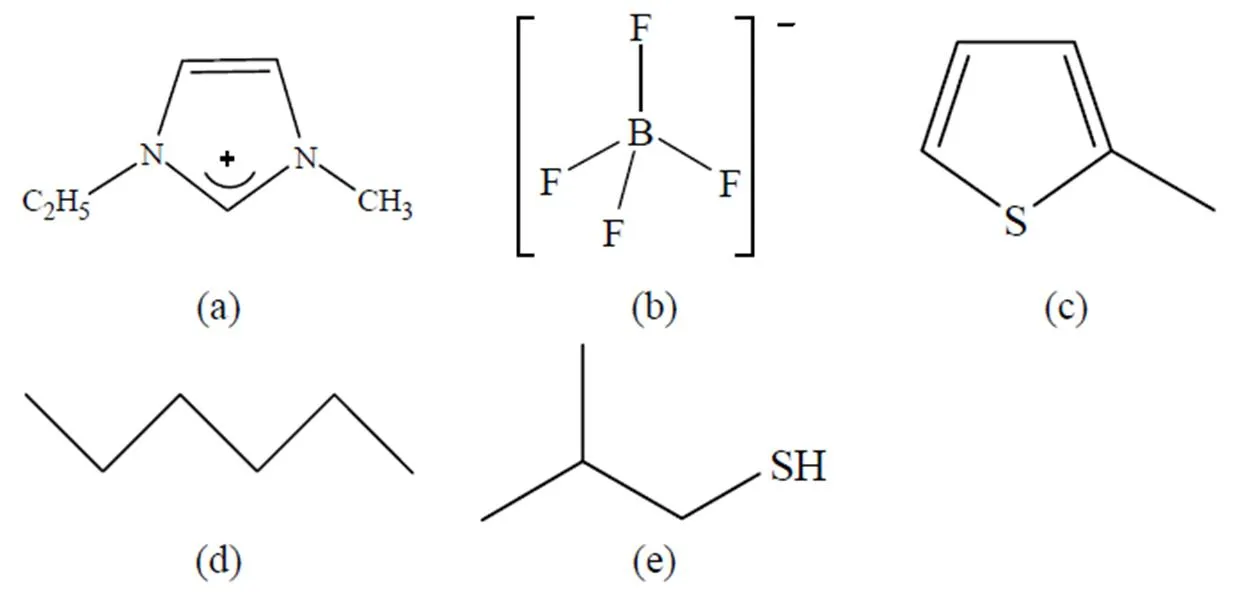
图1 (a) 1-乙基-3-甲基咪唑阳离子([EMIM]+) (b) 四氟合硼酸根阴离子[BF4]- (c) 2-甲基噻吩 (2-MT) (d) 正己烷 (HEX) 和 (e) 异丁基硫醇 (IBT)的结构
2 计算方法
密度泛函理论(DFT)比Hartree-Fock方法可以得到更可靠的氢键体系,并且所用优化时间远远小于MP2方法。密度泛函理论还成功的用于离子液体的优化。采用MS中DMol3软件包[18-19]中的GGA/PW91的泛函[20]和DNP基组对所有初始结构进行了非限制性全优化。尽管PW91泛函不能很好地描述色散作用,但GGA/PW91/DNP可以给出很好的优化结果[21]。最稳定结构的频率分析表明没有虚频。对最稳定的结构进行了NBO[22]和AIM[23-24]分析。
由于DMol3采用的是数字函数,比传统的Gaussian函数计算所得的基组重叠误差(BSSE)要小,所以可以忽略[25]。将[EMIM][BF4]-2-MT/HEX/ IBT的能量与[EMIM][BF4]、2-甲基噻吩/正己烷/异丁基硫醇能量差定义为相互作用能,其计算公式如下:
D= -{([EMIM][BF4]-2-MT/HEX/IBT) –
[([EMIM][BF4]) +(2-MT/ HEX/IBT)]}
3 结果与讨论
3.1 优化结构及电子性质
为了寻找到最稳定的[EMIM][BF4]结构,将[BF4]-阴离子分别置于咪唑环及乙基、甲基的周围,考虑到所有可能形成的氢键形式,如一般氢键、二叉氢键、三叉氢键。通过GGA/PW91/DNP全优化结果表明,如图2a所示的结构能量最低,最稳定。由图2a可以看出,[BF4]-阴离子位于咪唑环的C2-H2一侧,并且被乙基和甲基所包围。由H2形成的两个氢键H2···F1、H2···F2的键长最短,分别为2.185Å,2.052 Å。一个氢原子同时和两个供电体相互作用形成氢键,这称为二叉氢键或三中心氢键(bifurcated hydrogen bonding or three centered hydrogen bonding)[26],所以H2形成二叉氢键。其它相互作用为H81···F2(2.250 Å)、H61···F1(2.486 Å),其相互作用的距离小于它们的范德华半径之和(1.20 Å和1.47 Å)[27],而H71···F4、H71···F4的距离为2.705 Å、2.772 Å,略大于它们的范德华半径之和。
2-甲基噻吩与[EMIM][BF4]的相互作用的最稳定的结构如图2b所示。由图可见,[BF4]-阴离子位于咪唑环的C2-H2和乙基附近,而2-甲基噻吩处于咪唑环的甲基和[BF4]-阴离子之间。[EMIM]+和[BF4]-之间的相互作用分别为H2···F3(1.805 Å)、H61···F3(2.704 Å)、H71···F1(2.536 Å)、H61···F4 (2.425 Å),由H2形成的氢键,键长最短,除H61···F3的键长略大于它们的范德华半径之和(2.67 Å)外,其它氢键键长均小于它们的范德华半径之和。2-甲基噻吩与[EMIM][BF4]的相互作用分别为H3’···F2(2.438 Å)、H3’···F3(2.706 Å)、H62’···F1(3.034 Å)、H81···C4(2.727 Å)、H83···S1(3.190 Å)。H81和C4的距离小于它们的范德华半径(1.20 Å和1.70 Å)[27]之和,而H83和S1的距离略大于它们的范德华半径(1.20 Å和1.80 Å)[27]之和,这说明2-甲基噻吩与[EMIM][BF4]的相互作用不仅有H···F氢键,又有S1和C4参加的相互作用。
图2c所示为优化的最稳定的[EMIM][BF4]- HEX结构。可以看出,[BF4]-阴离子位于咪唑环上的C2-H2附近,并且能有效的和乙基、甲基发生作用,阴阳离子之间的相互作用分别为H81···F2(2.150 Å)、H2···F2(2.169 Å)、H2···F1(2.071 Å)、H71···F4(2.533 Å)、H71···F1(2.897 Å)、H61···F1(2.585 Å),其中只有H71···F1的距离略大于它们的范德华半径之和,其余的都小于它们的范德华半径之和。而正己烷与[EMIM][BF4]的相互作用是通过与[BF4]-阴离子中的氟原子相互作用实现的,它们相互作用的距离分别为3.288 Å(H11’···F1)、2.927 Å(H31’···F1)、2.857Å(H31’···F3)、和2.873 Å(H11’···F1),明显大于它们的范德华半径之和,这说明[EMIM][BF4]和正己烷的相互作用较弱。
异丁基硫醇与[EMIM][BF4]的相互作用的最稳定的结构如图2d所示。由图可见,[BF4]-阴离子位于[EMIM]+阳离子和异丁基硫醇的中间。[EMIM]+和[BF4]-的相互作用分别为H61···F4(2.656 Å)、H2···F4(1.932 Å)、H81···F1(2.373 Å)。[EMIM][BF4]与异丁基硫醇相互作用分别为H41’···F4(2.863 Å)、H32’···F4(2.737 Å)、H11’···F4(2.713Å)、H11’···F1(2.652 Å)、和H82···S(2.761 Å)。其中H和F之间的距离在范德华半径和(2.67 Å)[27]附近,而H和S的距离远远小于它们的范德华半径之和(3.00Å)[27],这说明S明显参见了异丁基硫醇与[EMIM][BF4]的相互作用。
在上述四个最稳定的结构中,由H2形成的氢键的距离最短,作用最大,这可能是由于咪唑环上的两个N原子的电负性相对较大,导致C2-H2上的正电荷较高,故而形成的氢键最强,这和文献报道是一致的[28]。
3.2 相互作用能
将[EMIM][BF4]-2-MT/HEX/IBT的能量与[EMIM][BF4]、2-甲基噻吩/正己烷/异丁基硫醇能量差定义为相互作用能。[EMIM][BF4]和2-甲基噻吩、正己烷、异丁基硫醇相互作用能分别为12.66 kcal/mol、5.21 kcal/mol和7.88 kcal/mol,说明相互作用为2-甲基噻吩 > 异丁基硫醇 > 正己烷,这与文献报道相一致[17]。


(a) (b) (c) (d)
3.3 NBO分析
采用B3LYP/6-31++G**方法对四个最稳定的优化结构进行了NBO分析。NBO分析可以提供分子中的原子集居数及分子间超共轭相互作用等。计算结果表明,在咪唑环上C2和H2的正电性远远高于C4、C5和H4、H5的正电性,这是由于N1和N3的电负性较大,吸引电子的能力较强,导致C2和H2的正电性明显高于其它原子上的正电性。因而由C2-H2形成的氢键最强。
为了研究弱相互作用的程度,表1所示为[EMIM][BF4]、[EMIM][BF4]-2-MT、[EMIM][BF4]- HEX和[EMIM][BF4]-IBT中的部分授-受相互作用及其二级微扰稳定化能E(2),E(2)数据表明电子授-受相互作用的强度。E(2)数据越大,电子授-受相互作用程度大,相互作用强。由表2所示,H2所形成的氢键作用最强,在[EMIM][BF4]中,LP(F2)®s*(C2-H2)的二级微扰稳定化能为4.80 kcal/mol,在[EMIM][BF4]-2-MT中,LP(F3)®s*(C2-H2) 的二级微扰稳定化能为18.01 kcal/mol,在[EMIM][BF4]- HEX中,LP(F1)®s*(C2-H2) 的二级微扰稳定化能为4.47 kcal/mol,在[EMIM][BF4]-IBT中,LP(F4)®s*(C2-H2) 的二级微扰稳定化能为16.02kcal/mol,这与它们距离最短是一致的。在[EMIM][BF4]中,其它相互作用分别为LP(F1)®s*(C2-H2)(2.20 kcal/mol)、LP(F4)®p*(N1-C2)(1.41 kcal/mol)、LP(F2)®s*(C8-H81)(2.20 kcal/mol)、LP(F1)®s*(C6-H61)(0.70 kcal/mol);在[EMIM] [BF4]-2-MT中,其它相互作用分别为LP(F1)®p*(N1-C2)(1.04 kcal/mol)、LP(F2)®s*(C3’-H3’) (1.44 kcal/mol)、LP(F1)®s*(C7-H71) (0.81 kcal/mol)、LP(F4)®s*(C6-H61)(1.07 kcal/mol);在[EMIM][BF4]-HEX 中,其它相互作用分别为s(F1-B)®s*(C2-H2)(0.74 kcal/mol)、LP(F2)®s*(C8-H81)(4.43 kcal/mol)、s(F2-B)®s*(C2-H2)(0.53 kcal/mol)、LP(F4)®p*(N1-C2)(0.97 kcal/mol)、LP(F4)®s*(C7-H71)(0.94 kcal/mol);在[EMIM][BF4]-IBT中,其它相互作用分别为LP(F3)®s*(C7-H73)(1.34 kcal/mol)、LP(F1)®s*(C8-H81)(1.38 kcal/mol)、LP(S)®s*(C8-H82)(2.64 kcal/mol)。
阴阳离子除静电作用外,四种最稳定的结构中主要相互作用为氢键作用,但是在[EMIM][BF4]- 2-MT中,不仅有氢键作用,还有p···H-C(p(C2’-C3’)®s*(C8-H81),p(C4’-C5’)®s*(C8-H81))的相互作用。在[EMIM][BF4]-IBT中,S明显参加了异丁基硫醇与[EMIM][BF4]的相互作用,这可能是由于在异丁基硫醇中,S的孤对电子没有参加共轭作用,易与s*(C8-H82)反键轨道发生较强作用;而在2-甲基噻吩中,由于噻吩环的共轭作用,导致2-甲基噻吩中的硫给出电子的能力较弱,所以其孤对电子与s*(C8-H83)、s*(N3-C8)反键轨道的相互作用较弱。

表1 [EMIM][BF4]、[EMIM][BF4]-2-MT、[EMIM][BF4]-HEX、[EMIM][BF4]-IBT中的部分授-受相互作用及其二级微扰稳定化能E(2) (kcal/mol)
3.4 拓扑性质分析
电子密度的拓扑学分析,主要是采用Bader[29]所提出的分子内原子(Atoms In Molecules,AIM)的理论。这个理论将量子力学所探究的观念应用到化学分子内部的电子密度,用来描述任何形式化学键的物理量。当Ñ2r(r)<0时,说明该临界点是极大值;当Ñ2r(r)>0时,说明该临界点是极小值;当Ñ2r(r) = 0时,代表该临界点为反曲点。若êÑ2r(r)ï值越大,说明该极值越明显[29]。
表2所示为3-MT、[EMIM][BF4]、[EMIM][BF4]-2-MT、[EMIM][BF4]-HEX和[EMIM][BF4]-IBT的电子密度(r)、Laplacian (Ñ2)和Hessian矩阵本征值(l1,l2,l3)。由表2所示,所有临界点的Ñ2BCP的值均为正值,这说明这些弱相互作用均为闭壳相互作用。[EMIM][BF4]中弱相互作用的拓扑性质分别为F2···H81 (= 0.01284 au,Ñ2= 0.04647 au)、F2···H2 (= 0.02036 au,Ñ2= 0.07091 au)、F4···H71 (= 0.00478 au,Ñ2= 0.02131 au)、F1···H2 (= 0.01558 au,Ñ2= 0.06203 au)、F1···H61 (= 0.00834 au,Ñ2= 0.00891 au)、F1···H71 (= 0.00482 au,Ñ2= 0.02215 au)。[EMIM][BF4]-2-MT中弱相互作用的拓扑性质分别为F2···H3’(= 0.00828 au,Ñ2= 0.03253 au)、F3···H3’(= 0.00525 au,Ñ2= 0.02410 au)、F3···H2 (= 0.03297 au,Ñ2= 0.10270 au)、F3···H61 (= 0.00527 au,Ñ2= 0.02424 au)、F1···H62’(= 0.00234 au,Ñ2= 0.01069 au)、F1···H71 (= 0.00724 au,Ñ2= 0.03080 au)、F4···H61 (= 0.00921 au,Ñ2= 0.03783 au)、C4’···H81 (= 0.00752 au,Ñ2= 0.02325 au)、S1···H83 (= 0.00515 au,Ñ2= 0.01725 au);[EMIM][BF4]-HEX中弱相互作用的拓扑性质分别为F2···H81 (= 0.01579 au,Ñ2= 0.05436 au)、F2···H2 (= 0.01613 au,Ñ2= 0.06302 au)、F4···H71 (= 0.00695 au,Ñ2= 0.02918 au)、F1···H2 (= 0.01949 au,Ñ2= 0.06869 au)、F1···H61 (= 0.00678 au,Ñ2= 0.02973 au)、F1···H71 (= 0.00375 au,Ñ2= 0.01750 au)、F1···H11’(= 0.00148 au,Ñ2= 0.00638 au)、F1···H31’(= 0.00308 au,Ñ2= 0.01369 au)、F3···H31’(= 0.00396 au,Ñ2= 0.01756 au)、F3···H51’(= 0.00348 au,Ñ2= 0.01572 au);[EMIM][BF4]-IBT中弱相互作用的拓扑性质分别为F4···H61 (= 0.00569 au,Ñ2= 0.02497 au)、F4···H32’(= 0.00462 au,Ñ2= 0.02014 au)、F4···H41’(= 0.00337 au,Ñ2= 0.01519 au)、F4···H2 (= 0.02584 au,Ñ2= 0.08030 au)、F4···H11’(= 0.00488 au,Ñ2= 0.02147 au)、F3···H73 (= 0.00962 au,Ñ2= 0.03794 au)、F1···H11’(= 0.00535 au,Ñ2= 0.02288 au)、F1···H81 (= 0.00989 au,Ñ2= 0.03979 au)、S···H82 (= 0.01068 au,Ñ2= 0.02898 au)。
由上述拓扑性质数据可以看出,在H···F相互作用中,其距离越短,则电子密度和Laplacian数据越大。并且在[EMIM][BF4]-2-MT中,出现了C4’···H81和S1···H83相互作用,C4’和H81的距离为2.727 Å,小于它们的范德华半径(1.70 Å 和1.20 Å)之和(2.90 Å),证明存在着p···H-C相互作用。尽管S1和H83的距离为3.190Å,略大于它们的范德华半径(1.80 Å和1.20 Å)之和(3.00 Å),但由于有明显的键临界点(BCP),所以存在着相互作用。在[EMIM][BF4]-IBT中,S···H82的距离为2.761 Å,远远小于它们的范德华半径之后,并且键临界点的电子密度和Laplacian数据较大,说明S形成的相互作用较强,说明异丁基硫醇上的硫给出电子的能力比2-甲基噻吩上的硫给出电子的能力要强,这与NBO分析结果相一致。比较环临界点(RCP)的电子密度和Laplacian数据可以看出,与小分子作用后,咪唑环的环临界点发生明显的变化,证明了彼此之间有相互作用。

表2 3-MT、[EMIM][BF4]、[EMIM][BF4]-2-MT、[EMIM][BF4]-HEX和[EMIM][BF4]-IBT的电子密度(r)、拉普拉斯密度(Ñ2r)和Hessian矩阵本征值(l1, l2, l3)
续表2 3-MT、[EMIM][BF4]、[EMIM][BF4]-2-MT、[EMIM][BF4]-HEX和[EMIM][BF4]-IBT的电子密度(r)、拉普拉斯密度(Ñ2r)和Hessian矩阵本征值(l1,l2,l3)
Table 2 The topological properties of electron density (r), Laplacian of density (Ñ2r), eigenvalues of the Hessian matrix (l1,l2,l3) of 3-MT, [EMIM][BF4], [EMIM][BF4]-2-MT, [EMIM][BF4]-HEX, [EMIM][BF4]-IBT (atomic units)
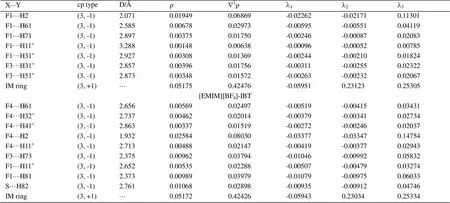
X×××Ycp typeD/ÅrÑ2rl1l2l3 F1···H2(3, -1)2.0710.019490.06869-0.02262-0.021710.11301 F1···H61(3, -1)2.5850.006780.02973-0.00595-0.005510.04119 F1···H71(3, -1)2.8970.003750.01750-0.00246-0.000870.02083 F1···H11’(3, -1)3.2880.001480.00638-0.00096-0.000520.00785 F1···H31’(3, -1)2.9270.003080.01369-0.00244-0.002100.01824 F3···H31’(3, -1)2.8570.003960.01756-0.00311-0.002550.02322 F3···H51’(3, -1)2.8730.003480.01572-0.00263-0.002320.02067 IM ring(3, +1)···0.051750.42476-0.059510.231230.25305 [EMIM][BF4]-IBT F4···H61(3, -1)2.6560.005690.02497-0.00519-0.004150.03431 F4···H32’(3, -1)2.7370.004620.02014-0.00379-0.003410.02734 F4···H41’(3, -1)2.8630.003370.01519-0.00272-0.002460.02037 F4···H2(3, -1)1.9320.025840.08030-0.03377-0.033470.14754 F4···H11’(3, -1)2.7130.004880.02147-0.00419-0.003770.02943 F3···H73(3, -1)2.3750.009620.03794-0.01046-0.009920.05832 F1···H11’(3, -1)2.6520.005350.02288-0.00507-0.004790.03274 F1···H81(3, -1)2.3730.009890.03979-0.01079-0.009750.06033 S···H82(3, -1)2.7610.010680.02898-0.00935-0.009120.04746 IM ring(3, +1)···0.051720.42426-0.059430.230340.25334
AIM理论不仅能表明弱相互作用的本性,还能通过电子密度的大小反映氢键的强度。电子密度越大,氢键键长越短,氢键越强。研究表明[30-31],H···O的距离与它们的键临界点的电子密度有一定的关系。所以我们考察了F···H的距离与它们的键临界点的电子密度对数的关系。图3所示d(F···H)与 ln(b)的关系。由图3可见,[EMIM][BF4]、[EMIM][BF4]-2-MT、 [EMIM][BF4]-HEX、[EMIM] [BF4]-IBT的相关因子分别为0.996、0998、0.998和0.999,这说明氢键键长与其强度有很好的相关性,可以通过电子密度估计氢键的强度。

(a)(b) (c)(d)
4 结论
采用GGA/PW91/DNP方法非限制性的全优化了[EMIM][BF4]、[EMIM][BF4]-2-MT、 [EMIM][BF4]-HEX、[EMIM][BF4]-IBT的初始结构。对最稳定的结构进行了NBO和AIM分析。[EMIM][BF4]、[EMIM][BF4]-2-MT、[EMIM][BF4]- HEX、[EMIM][BF4]-IBT的最稳定结构显示[BF4]-阴离子总是优先靠近咪唑基中的C2-H2的附近,这是由于咪唑环上N1和N3的电负性较大,造成C2-H2的正电性较高。2-甲基噻吩、正己烷、异丁基硫醇与[EMIM][BF4]相互作用并没有改变[BF4]-阴离子与C2-H2的优先作用。氢键在[EMIM][BF4]与正己烷、异丁基硫醇的相互作用中起重要作用,而[EMIM][BF4]与2-甲基噻吩存在着氢键和p···C-H相互作用。[EMIM][BF4]与2-甲基噻吩、正己烷、异丁基硫醇的相互作用能分别为12.66 kcal/mol、5.21 kcal/mol和7.88 kcal/mol,说明[EMIM][BF4]会优先吸附2-甲基噻吩。
[1] Bosmann A, Datsevich L, Jess A, et al.Deep desulfurization of diesel fuel by extraction with ionic liquids[J]. Chem. Commun., 2001, 2494-2495.
[2] Lo W, Yang HWei G. One-pot desulfurization of light oils by chemical oxidation and solvent extraction with room temperature ionic liquids[J]. Green Chem., 2003(5): 639-642.
[3] 李红海,李婧,王伟文,等,离子液体脱硫性能研究与应用进展[J].化学工业与工程,2012,29:73-79.
[4] 高红帅,李望良,邢建民,等.离子液体用于燃料油深度脱硫的研究进展[J].石油化工,2007,36:966-970.
[5] Li H, He L, Lu J, et al. Deep oxidative desulfurization of fuels catalyzed by phosphotungstic acid in ionic liquids at room temperature[J]. Energy Fuels,2009, 23: 1354-1357.
[6] Lissner E, de Souza W F, Ferrera B, et al. Oxidative desulfurization of fuels with task-specific ionic liquids[J]. ChemSusChem, 2009(2): 962-964.
[7] Zhao D, Wang Y, Duan E, et al. Oxidation desulfurization of fuel using pyridinium-based ionic liquids as phase-transfer catalysts[J]. Fuel Processing Technol., 2010, 91: 1803-1806.
[8] Seeberger A., Jess A.. Desulfurization of diesel oil by selective oxidation and extraction of sulfur compounds by ionic liquids—A contribution to a competitive process design[J].Green Chem, 2010, 12: 602-608.
[9] Zhang W, Xu K, Zhang Q, et al.Oxidative desulfurization of dibenzothiophene catalyzed by ionic liquid [BMIm]HSO4[J].Ind. Eng. Chem. Res.,2010, 49: 11760- 11763.
[10] Ding Yuxiao, Zhu Wenshuai, Li Huaming, et al. Catalytic oxidative desulfurization with a hexatungstate/aqueous H2O2/ionic liquid emulsion system[J].Green Chem, 2011, 13: 1210-1216.
[11] Hu Y, He Q, Zhang Z, et al. Oxidative desulfurization of dibenzothiophene with hydrogen peroxide catalyzed by selenium (IV)-containing peroxotungstate[J].Chem. Commun.,2011. 12194-12196.
[12] 佟拉嘎,李巍,荣华,等.缬氨酸阳离子型离子液体的实验与理论研究[J].高等学校化学学报,2011,32:927-933.
[13] Kumar A A P, Banerjee T. Thiophene separation with ionic liquids for desulphurization: A quantum chemical approach[J]. Fluid Phase Equilib., 2009, 278: 1-8.
[14] Anantharaj R, Banerjee T. Quantum chemical studies on the simultaneous interaction of thiophene and pyridine with ionic liquids[J]. AIChE J., 2011, 57: 749-764.
[15] Lü R, Qu Z, Yu H, et al. Comparative study on interactions between ionic liquids and pyridine/hexane[J]. Chem. Phys. Lett., 2012, 532: 13-18.
[16] Lü R, Qu Z, Yu H, et al. The electronic and topological properties of interactions between 1-butyl-3- methylimidazolium hexafluorophosphate/tetrafluoro borate and thiophene[J]. J. Mol. Graph. Model., 2012, 36:36-41.
[17] Zhang S, Zhang Z C. Novel properties of ionic liquids in selective sulfur removal form fuels at room temperature[J]. Green Chem., 2002(4): 376-379.
[18] Delley B. An all-electron numerical method for solving the local density functional for polyatomic molecules[J].J. Chem. Phys., 1990, 92, 508-517.
[19] Delley B. From molecules to solids with the DMol3approach[J].J. Chem. Phys., 2000, 113: 7756-7764.
[20] Perdew J P, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy[J]. Phys. Rev. B, 1992, 45:13244-13249.
[21] Castellano O, Gimon R, Soscun H. Theoretical study of thes-pandp-pinteractions in heteroaromatic monocyclic molecular complexes of benzene, pyridine, and thiophene dimers: Implications on the resin- asphaltene stability in [J]. Energy Fuel,2011,25:2526-2541.
[22] Reed A E, Curtiss L A, Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint[J]. Chem. Rev., 1988, 88: 899-926.
[23] Biegler-KönigF, Schönbohm J. Update of the AIM2000 program for atoms in molecules[J]. J. Comput. Chem., 2002, 23: 1489-1494.
[24] Biegler-König F, Schönbohm J, Bayles D. AIM2000 - A program to analyze and visualize atoms in molecules[J]. J. Comput. Chem., 2001, 22: 545-559.
[25] Inada Y, Orita H.Efficiency of numerical basis sets for predicting the binding energies of hydrogen bonded complexes: Evidence of small basis set superposition error compared to Gaussian basis sets[J]. J. Comput. Chem., 2008, 29:225-232.
[26] Rozas I, Alkorta I, Elguero J. Bifurcated hydrogen bonds: three-centered interactions[J]. J. Phys. Chem. A, 1998, 102: 9925-9932.
[27] Bondi A. van der Waals volumes and radii[J]. J. Phys. Chem., 1964, 68: 441-451.
[28] Hunt P A, Kirchner B, Welton T. Characterising the electronic structure of ionic liquids: An examination of the 1-butyl-3-methylimidazolium chloride ion pair[J]. Chem. Eur. J., 2006, 12: 6762-6775.
[29] Bader R F W. A quantum theory of molecular structure and its applications[J]. Chem. Rev., 1991, 91: 893-928.
[30] Espinosa E, Souhassou M, Lachekar H, et al. Topological analysis of the electron density in hydrogen bonds[J]. Acta Cryst., 1999, 55: 563-574.
[31] Netzel J, van Smaalen S. Topological properties of hydrogen bonds and covalent bonds from charge densities obtained by the maximum entropy method (MEM) [J]. Act Cryst., 2009, 65: 624-638.
THEORETICAL STUDY ON INTERACTION BETWEEN IMIDAZOLYL IONIC LIQUIDS AND SMALL MOLECULES
LIN Jin1,*Lü Ren-qing2
(1. College of Chemical Engineering, China University of Petroleum (East China), Qingdao, Shandong 266580, China; 2. College of Science, China University of Petroleum (East China), Qingdao, Shandong 266580, China)
Density functional theory has been employed to investigate the interaction between 2-methyl-thiophene (2-MT), hexane (HEX), isobutylthiol (IBT) and1-ethyl-3-methylimidazolium tetrafluoroborate ([EMIM][BF4]). GGA/PW91 functionals and DNP basis set were used to optimize the geometries, and the hydrogen bond interaction between [EMIM][BF4] and 2-methyl-thiophene (2-MT), hexane (HEX), isobutylthiol (IBT) have been analyzed by NBO and AIM methods. The most stable gas-phase structure of [EMIM][BF4] ion pair indicates that hydrogen bond interaction between the fluorine atoms on [BF4]-anions and the C2-hydrogen on the imidazole ring plays an important role in the formation of ion pair. Additional interaction is observed between [BF4]-anion and the hydrogen atoms on the adjacent alkyl side chains of [EMIM]+cation. The [BF4]-anion tends to C2-H2 forming hydrogen bond, suggesting that 2-methyl-thiophene (2-MT), hexane (HEX), isobutylthiol (IBT)’s adsorption on [EMIM][BF4] do not change the dominant role of ionic liquids pair. Thep···C-H interaction and hydrogen bonding interaction occur between [EMIM][BF4] and 2-methyl-thiophene. The hydrogen bonding interaction exists between [EMIM][BF4] and hexane, isobutylthiol. The interaction energy suggests that 2-methyl-thiophene is prior to adsorpt on ionic liquids.
ionic liquid; density functional theory; 2-methyl-thiophene; hexane; isobutylthiol
O641
A
10.3969/j.issn.1674-8085.2013.01.009
1674-8085(2013)01-0039-08
2012-11-18;
2012-12-06
林 进(1990-),男,江西井冈山人,中国石油大学(华东)化学工程学院本科生 (E-mail: 1311870044@qq.com)
*吕仁庆(1969-),男,山东莱阳人,副教授,博士,主要从事离子液体及脱硫脱氮的理论研究(E-mail: lvrenqing@upc.edu.cn).
