高效液相色谱法同时测定盆炎净片中原儿茶酸、咖啡酸和芍药苷含量
2012-06-27张晓娟
黎 强 ,张晓娟 ,罗 玲
盆炎净片由忍冬藤、狗脊、蒲公英、益母草、车前草、川芎、赤芍、鸡血藤8味中药组方,具有清热利湿、利血通络、调经止带功效[1]。方中忍冬藤清热解毒,疏风通络;鸡血藤主要补血,活血通络;狗脊补肝肾,强腰膝;益母草活血调经,利尿消肿;川芎活血行气,祛风止痛;赤芍清热凉血,散瘀止痛。原标准仅选取忍冬藤及赤勺作为定性指标,赤勺中芍药苷作为定量指标。有学者增加鸡血藤、益母草、川芎的鉴别作为定性指标,增加忍冬藤中绿原酸的含量测定,提高了盆炎净片的质量控制要求[2]。为更全面地控制药品质量,本试验中拟用高效液相色谱(HPLC)法同时测定盆炎净片中原儿茶酸、咖啡酸、芍药苷的含量,现报道如下。
1 仪器与试药
Agilent 1100型Series高效液相色谱仪(四元泵,在线脱气机,自动进样器,柱温箱);TU-1221型紫外可见分光光度计(北京普析通用);AE240型电子分析天平(梅特勒-托利多);SB2200型超声清洗器(上海必能信超声有限公司)。乙腈、磷酸(色谱纯),纯净水(杭州娃哈哈集团);原儿茶酸对照品(批号为809-200102),咖啡酸对照品(批号为110885-200102),芍药苷对照品(批号为110736-200934,含量95.7%),均购于中国药品生物制品检定所,供含量测定用;盆炎净片(陕西白鹿制药股份有限公司);忍冬藤、鸡血藤、蒲公英、狗脊、川芎、益母草、车前草、赤勺等8味药材均为市售品。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Agilent Eclipse XDB-C18柱(250 mm×4.6 mm,5μm);流动相:乙腈-0.1%磷酸梯度洗脱,0~10 min时乙腈 8%和0.1%磷酸 92%,10~25 min时乙腈 14%和 0.1%磷酸 86%;流速:0.8 mL/min;检测波长:219 nm;进样量:10 μL。在此条件下,3个组分与相邻色谱峰的分离度均大于1.5,理论板数按原儿茶酸、咖啡酸、芍药苷计均大于5 000(图1)。
2.2 溶液制备
取盆炎净片10片,研细,取约0.5 g,精密称定,置具塞锥形瓶中,精密加入15 mL甲醇,称定质量,超声处理20 min,放冷,用甲醇补足质量,摇匀,过滤,用0.45μm微孔滤膜滤过,取续滤液作为供试品溶液。精密称取原儿茶酸、咖啡酸和芍药苷对照品适量,以甲醇为溶剂分别制成质量浓度为 1.4,1.4,0.6 g/L 的贮备液,并制备含原儿茶酸28μg/mL、咖啡酸28μg/mL和芍药苷120μg/mL的混合对照品溶液。
2.3 方法学考察
专属性试验:按处方制备分别不含鸡血藤、蒲公英及赤勺原药材的样品,按供试品溶液的制备方法制备阴性对照品溶液,依法进样。结果分别在原儿茶酸、咖啡酸和芍药苷的对应峰位上未出现色谱峰,说明阴性对照品溶液对测定无干扰(见图1)。
线性关系考察:分别精密吸取对照品贮备液,采用等量递减方法制得系列质量浓度的混合对照品溶液,进样分析,记录色谱峰面积,以对照品质量浓度(X)为横坐标、峰面积(Y)为纵坐标绘制标准曲线。结果见表1。
精密度试验:将同一混合对照品溶液在上述色谱条件下重复进样5次。结果原儿茶酸、咖啡酸和芍药苷峰面积的 RSD分别为0.7% ,0.4%,0.5%。

图1 高效液相色谱图
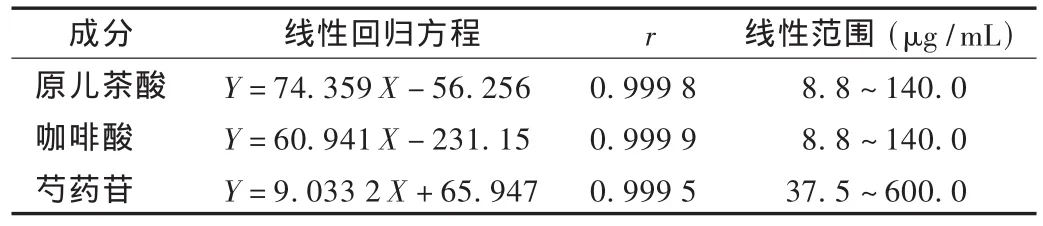
表1 线性关系考察结果
稳定性试验:取同一供试品溶液,分别在 0,2,4,8,12 h时按上述色谱条件进行测定。结果原儿茶酸、咖啡酸和芍药苷峰面积的 RSD分别为0.8%,1.8%,1.0%,表明供试品溶液至少在12 h内稳定。
重复性试验:取同一批样品(批号为100306),依法平行制备6份供试品溶液并进样分析。结果原儿茶酸、咖啡酸和芍药苷含量平均值为 1.605 9,1.556 9,3.655 2 mg/g,RSD 分别为 1.2%,1.4% ,1.0%。
加样回收试验:分别取已知含量的样品(批号为100306)6份,每份约0.25 g,精密称定,分别精密加入对照品贮备液,挥干溶剂后按供试品溶液制备方法制备溶液并测定各成分的含量,计算回收率。结果见表2。

表2 原儿茶酸、咖啡酸、芍药苷加样回收试验结果(n=6)
2.4 样品含量测定
取3批样品,依法制备供试品溶液并进样测定,以外标法计算含量。结果见表3。

表3 样品含量测定结果(mg/g)
3 讨论
将适当质量浓度的原儿茶酸、咖啡酸和芍药苷对照品溶液在200~400 nm波长范围内进行吸收光谱扫描,结果原儿茶酸在259 nm和219 nm波长处有最大吸收,咖啡酸在323 nm和219 nm波长处有最大吸收,芍药苷在229 nm波长处有最大吸收,经综合考量,最后确定测定波长为219 nm。
因本试验中成分靠近末端吸收,在色谱条件筛选试验中,比较乙腈-水、乙腈-1%磷酸、乙腈-0.1%磷酸溶液3个流动相后,确定乙腈-0.1%磷酸为最终条件。当进样量为20μL时,样品过载,峰形严重前沿,而调整进样量为10μL后,则样品峰形尖锐,对称性良好。
考察使用溶剂时,曾采用甲醇、50%甲醇、乙醇对样品进行提取,结果甲醇的提取效率最高;后通过超声15,20,30 min,回流1,1.5 h,振荡等提取方法比较,得出用甲醇超声提取20 min时提取效果最好,基线相对较平[5-6]。
含量测定中,不同批号样品间咖啡酸含量差别较大。而咖啡酸会与奎尼酸反应生成绿原酸[7],是否因此原因导致咖啡酸含量不稳定,仍需要进一步验证。
[1]YBZ25272005,国家食品药品监督管理局标准(试行)[S].
[2]杨遁嘉,霍 昕,刘文炜,等.鸡血藤颗粒质量标准的研究[J].中成药,2009,3(1):144 -146.
[3]尹 伟,岳春华,雷 伟,等.盆炎净片的质量控制[J].中南药学,2007,5(3):246 - 149.
[4]钱正明,李会军,李 萍.高效液相色谱法测定忍冬藤和叶中8种活性成分[J].分析化学,2007,8(35):1 159 -1 163.
[5]邓君丽,王兆霞.HPLC测定消炎片中咖啡酸的含量[J].中成药,2007,29(4):615 -616.
[6]耿家玲,盂 芹.HPLC法测定灯盏细辛中绿原酸和咖啡酸的含量[J].中国药师,2010,13(5):701 -702.
[7]孟 伟,温亚男,魏永巨,等.金银花中绿原酸含量的荧光法测定[J].分析化学,2009,37(A01):211.
