健儿消食颗粒质量标准研究
2012-06-27韩云霞靳凤云李德鑫
韩云霞,李 平,靳凤云,李德鑫
健儿消食颗粒由黄芪、白术、陈皮、麦冬、黄芩、山楂、莱菔子7味中药组方,具有健脾益胃、理气消食之功效,可用于小儿饮食不节损伤脾胃引起的纳呆食少、脘胀腹满、自汗乏力、大便不调,以至厌食、恶食等症[1]。该制剂由口服液改剂型而来,原质量控制标准没有含量测定项目。为更好地控制该制剂质量,本研究中对制剂中黄芩、黄芪、莱菔子、陈皮等4味药进行薄层定性鉴别研究,以黄芩中有效成分黄芩苷进行了含量测定研究。现报道如下。
1 仪器与试药
LC-10ATvp泵,SPD-10Avp紫外检测器,CTO-10Asvp柱温箱(日本岛津);Shimadzu Libror AEL 40SM(十万分之一)天平(日本岛津);硅胶G薄层板(青岛洋化工厂)。健儿消食颗粒(贵阳中医学院第二附属医院制剂室,批号分别为20080901,20080902,20080903);黄芩对照药材(批号为 120955 -200301),没食子酸对照品(批号为110830-200301),莱菔子对照药材(批号 为 121074-200302),黄芪 对照 药材(批 号为 120927-200310),陈皮苷对照品(批号为0775-9901),黄芩苷对照品(批号为110715-200212),购自中国药品生物制品检定所;乙腈(天津市大茂化学试剂厂),其他试剂均为分析纯,水(重蒸馏,临用前制备)。
2 方法与结果
2.1 薄层色谱鉴别
黄芪:取本品10 g,研细,加乙醇40 mL,超声处理1 h,滤过,滤液浓缩至约2 mL,作为供试品溶液。取不含黄芪的阴性样品,同法制备阴性对照品溶液。另取黄芪对照药材2 g,加乙醇10 mL,同法制成对照药材溶液。照薄层色谱法[2005年版《中国药典(一部)》附录ⅥB]试验,吸取上述3种溶液各10μL,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以苯-醋酸乙酯(8∶2)为展开剂,展开,取出,晾干,置氨蒸气饱和的层析缸内熏5 min后,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照药材溶液色谱相应的位置上,显相同颜色的斑点,见图1 A。
黄芩:取本品 10 g,加水 50 mL,煮沸 10 min,放冷,滤过,滤液加乙醚提取2次,每次25 mL,分取乙醚层,挥至约2 mL,作为供试品溶液。取不含黄芩的阴性样品,同法制备阴性对照品溶液。另取黄芩对照药材1 g,同法制成对照药材溶液。照薄层色谱法[2005年版《中国药典(一部)》附录ⅥB]试验,吸取上述3种溶液各10μL,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以氯仿-甲醇(9∶1)为展开剂,展开,取出,晾干,以碘蒸气熏至斑点显色清晰。供试品溶液色谱中,在与对照药材溶液色谱相应的位置上,显相同颜色的斑点,见图1 B。
山楂:取本品10 g,研细,加乙酸乙酯25 mL,超声处理30 min,滤过,滤液挥至约1 mL,作为供试品溶液。取不含山楂的阴性样品,同法制备阴性对照品溶液。另取山楂对照药材1 g,加乙酸乙酯5 mL,超声处理15 min,滤过,滤液作为对照药材溶液。照薄层色谱法[2005年版《中国药典(一部)》附录ⅥB]试验,吸取上述3种溶液各10μL,分别点于同一硅胶G薄层板上,以甲苯-醋酸乙酯-甲酸(20∶4∶0.5)为展开剂,展开,取出,晾干,喷以硫酸乙醇溶液(3→10),在80℃加热至斑点显色清晰。供试品溶液色谱中,在与对照药材溶液色谱相应的位置上,显相同颜色的斑点,见图1 C。
陈皮:取本品10 g,研细,加甲醇25 mL,超声处理30 min,滤过,滤液蒸干,残渣加甲醇2 mL溶解,取上清液作为供试品溶液。取不含陈皮的阴性样品,同法制备阴性对照品溶液。另取陈皮对照药材1 g,加甲醇5 mL,超声处理30 min,滤过,滤液挥至约1 mL,作为对照药材溶液。照薄层色谱法[2005年版《中国药典(一部)》附录ⅥB]试验,吸取上述3种溶液各10μL,分别点于同一用1%羧甲基纤维素钠溶液制备的硅胶G薄层板上,以醋酸乙酯-甲醇-水(100∶17∶13)为展开剂,展至约3 cm,取出,晾干,再以喷以三氯化铝试液,置紫外光灯(365 nm)下检视。供试品溶液色谱中,在与对照药材溶液色谱相应的位置上,显相同颜色的荧光斑点,见图1 D。

图1 薄层色谱图
2.2 含量测定
2.2.1 测定波长选择
取黄芩苷对照品适量,配制成对照品溶液,置分光光度计中,于190~400 nm波长范围内扫描,结果黄芩苷对照品溶液在280 nm处有最大吸收。经验证,供试品在此条件下测定效果良好,故选定280 nm为黄芩苷的检测波长。
2.2.2 色谱条件
色谱柱:Diamonsil C18柱(250 mm ×4.6 mm,5μm);流动相:乙腈 -水 -磷酸(30∶70∶0.04);流速:1 mL/min;柱温:35℃;进样量:10μL;检测波长:280 nm。
2.2.3 溶液制备
精密称取经60℃减压干燥4 h的黄芩苷对照品适量,置棕色容量瓶中,加甲醇使溶解,摇匀,制成每1 mL含黄芩苷40μg的溶液,即得对照品溶液。取装量差异项下的本品约0.5 g,研细,精密称定,置100 mL具塞锥形瓶中,精密加入70%乙醇25 mL,称定质量,超声提取(功率 260 W,频率 40 kHZ)30 min,取出,放冷,称定质量,用70%的乙醇补足减失质量,摇匀,用微孔滤膜(0.45μm)滤过,取续滤液,即得供试品溶液。取不含黄芩的阴性样品,同供试品溶液制备方法制备阴性对照品溶液。
2.2.4 方法学考察
系统适用性试验:分别取对照品溶液、供试品溶液、阴性对照品溶液各10μL注入色谱仪,记录色谱图,见图2。阴性对照品溶液色谱图在黄芩苷峰位置无色谱峰,表明其他组分对测定无干扰。黄芩苷峰与相邻组分峰的分离度在于1.5,理论板数以黄芩苷峰计不小于5 000。
线性关系考察:精密称取黄芩苷对照品12.19 mg,置25 mL量瓶中,加甲醇溶解并稀释至刻度,摇匀,精密吸取2 mL,置25 mL量瓶中,加甲醇稀释至刻度,得质量浓度为0.039 01 g/L的黄芩苷对照品溶液,分别精密吸取 2.5,5.0,10.0,15.0,20.0 μL,注入色谱仪,记录色谱图,测定峰面积积分值,以对照品进样量(μg)为横坐标、峰面积值为纵坐标绘制标准曲线,得回归方程 Y=1 938.29 X -5.88,r=0.999 9(n=5)。结果表明,黄芩苷进样量在0.098~0.780μg范围内与峰面积线性关系良好。
稳定性试验:取同一供试品(批号为20080901)溶液,在室温下固定时间0,2,4,8,12 h进样测定黄芩苷峰面积值。结果的RSD=0.85%(n=5),表明黄芩苷在12 h内基本稳定。
精密度试验:精密吸取对照品溶液(质量浓度为0.04184g/L),重复进样5次,测定黄芩苷峰面积积分值。结果的 RSD=0.9%(n=5),表明仪器精密度良好。
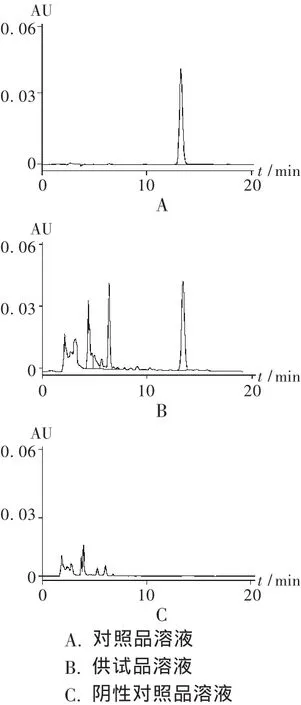
图2 高效液相色谱图
重复性试验:精密取本品内容物(批号为20080901)各0.5 g,共5份,依法制备供试品溶液并测定含量。结果的 RSD=0.96%(n=5),表明方法重现性良好。
加样回收试验:称取已知含量的同一批样品约0.25 g(批号为20080901,黄芩苷含量为2.01 mg/g)6份,分别加入黄芩苷对照品溶液(0.541 g/L)1 mL,按供试品溶液制备方法制备待测溶液,依法测定含量。结果见表1。

表1 黄芩苷加样回收试验结果(n=6)
2.2.5 样品含量测定
分别取10批样品内容物适量,按质量标准草案方法测定其中黄芩苷含量。结果见表2。

表2 10批样品中黄芩苷的含量测定结果(mg/袋)
3 讨论
在薄层鉴别研究中,笔者曾对方中白术、麦冬、莱菔子3味药材分别采用了醋酸乙酯-甲酸-水、石油醚-醋酸乙酯、甲苯-甲醇-冰醋酸等多种类、不同比例展开剂反复试验,但因分离效果不好、重现性差及阴性干扰等原因而未列入质量标准正文。
试验中考察了不同提取方法、不同提取溶剂、不同提取时间。以超声处理和热回流提取时,结果两者黄芩苷的提取量较相近,从操作方便考虑,选择超声提取;以甲醇、乙醇、70%乙醇为溶剂超声提取,结果表明以70%乙醇为溶剂黄芩苷提取量最高;以70%乙醇为溶剂超声15,30,45 min,结果表明,供试品中的黄芩苷在30~45 min时含量变化已不大,说明黄芩苷基本提尽,故选30 min为提取时间。
本制剂由7味中药材组方,本试验对4味药材进行薄层色谱定性鉴别,同时还对黄芩进行含量测定,此质量控制方法能很好控制产品质量。
[1]WS3-B-0997-91,中华人民共和国卫生部药品标准·中药成方制剂(第五册)[S].
