HPLC法同时测定痰热清注射液中木犀草苷和黄芩苷的含量Δ
2011-11-09姚文冰广西北海食品药品检验所北海市536000
姚文冰(广西北海食品药品检验所,北海市 536000)
痰热清注射液是国家中药二类新药,由金银花、黄芩、连翘等5味药材经提取加工制成,具有抑菌、抗病毒等功效[1],主要用于小儿上呼吸道感染和急性肺炎的防治。目前,未见同一色谱条件下检测金银花和黄芩含量方法的文献报道,故笔者通过相应的方法学考察建立了以高效液相色谱(HPLC)法同时测定痰热清注射液中木犀草苷、黄芩苷2个指标性成分含量的方法。结果表明,所建方法简单、快速、准确,可有效用于痰热清注射液的质量控制。
1 仪器与试药
1200型HPLC仪(美国Agilent公司);CP225D型电子天平(德国Sartorius公司);AW-120型电子天平(西班牙COBOS公司);SB3200S超声波清洗器(必能信超声(上海)有限公司,功率:250 W,频率:40 kHz)。
木犀草苷、黄芩苷对照品(中国药品生物制品检定所,批号分别为110795-200806、110715-200514);痰热清注射液(上海市某厂,批号:20101214、20100711、20100121);乙腈为色谱纯,其他试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:Agilent ZORBAX SB-phenyl(250 mm×4.6 mm,5 μm);流动相:乙腈(A)-0.5%冰醋酸(B),梯度洗脱(0→30 min,10%A→70%A;30→40 min,70%A→30%A;40→45 min,30%A→10%A);检测波长:350 nm;流速:0.8 mL·min-1;柱温:室温;进样量:供试品30µL、对照品10µL。
2.2 溶液的制备
2.2.1 混合对照品贮备液 精密称取经五氧化二磷减压干燥12 h以上的木犀草苷、黄芩苷对照品各适量,加入50%甲醇制成木犀草苷和黄芩苷浓度分别为40.92、19.72µg·mL-1的混合对照品贮备液,即得。
2.2.2 供试品溶液 精密量取样品20 mL,置分液漏斗中,用乙酸乙酯提取5次,每次25 mL,合并乙酸乙酯液,蒸干,残渣用50%甲醇使溶解并转移至5 mL量瓶中,加50%甲醇至刻度,摇匀,用微孔滤膜(0.45 μm)滤过,即得。
2.2.3 阴性对照溶液 分别取按处方工艺制备的不含金银花、黄芩的阴性样品,照“2.2.2”项下方法制得阴性对照溶液。
2.3 专属性考察
分别吸取上述混合对照品贮备液10µL,供试品溶液、阴性对照溶液各30µL,按“2.1”项下色谱条件进样检测。结果,供试品溶液中木犀草苷、黄芩苷与杂质峰完全分离,阴性对照无干扰。色谱见图1。

图1 高效液相色谱图A.混合对照品;B.供试品;C.阴性对照;1.木犀草苷;2.黄芩苷Fig1 HPLC chromatorgramsA.mixed reference substance;B.test sample;C.negative control;1.luteolin;2.baicalin
2.4 线性关系考察
取上述混合对照品贮备液适量,加50%甲醇制成木犀草苷、黄芩苷浓度分别为0.16、0.79µg·mL-1的混合对照品溶液,依次自动进样5、10、15、20、25 µL,记录峰面积。以峰面积积分值(Y)对进样量(X,µg)进行线性回归,得木犀草苷的回归方程为Y=38.786X+0.2(r=0.999 3,n=5),线性范围为0.82~4.09µg;黄芩苷的回归方程为Y=104.59X-54.037(r=0.999 6,n=5),线性范围为2.37~19.72 µg。
2.5 精密度试验
精密吸取混合对照品贮备液10µL,按“2.1”项下色谱条件连续进样6次,记录峰面积。结果,木犀草苷、黄芩苷的RSD分别为0.20%、0.92%(n=6),表明仪器精密度良好。
2.6 稳定性试验
精密吸取同一供试品溶液适量,按“2.1”项下色谱条件于室温下0、2、4、6、8、12、24 h各进样30 µL,记录峰面积。结果,木犀草苷、黄芩苷的RSD分别为1.24%、0.56%(n=6),表明供试品溶液在24 h内基本稳定。
2.7 重复性试验
取痰热清注射液(批号:20100711)适量,按“2.2.2”项下方法平行制备6份供试品溶液,分别进样测定。结果,木犀草苷、黄芩苷的平均含量分别为1.87、8.25µg·mL-1,RSD分别为1.46%、0.21%(n=6),表明本方法重复性良好。
2.8 加样回收率试验
精密量取已知含量的样品(批号:20100711)10 mL,加入适量的木犀草苷、黄芩苷对照品,按“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件测定,计算加样回收率,结果见表1。
2.9 样品含量测定
取3批痰热清注射液适量,分别按“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件测定样品含量,结果见表2。
3 讨论
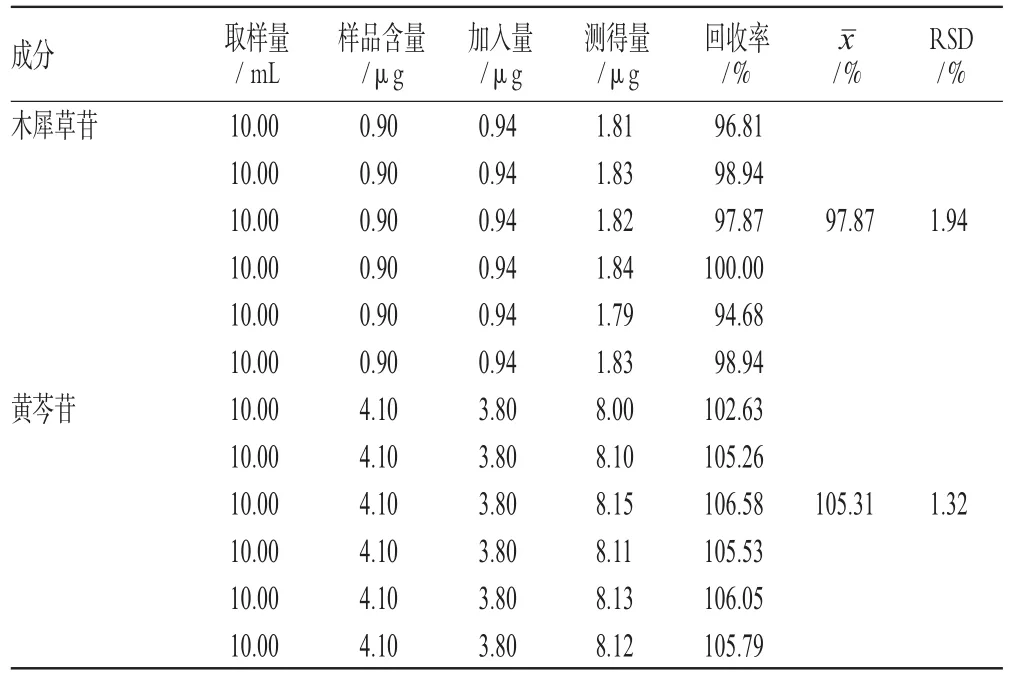
表1 加样回收率试验结果(n=6)Tab 1Results of recovery tests(n=6)

表2 样品含量测定结果(µg·mL-1,n=3)Tab2 Results of content determination of samples(µg·mL-1,n=3)
3.1 检测波长的选择
黄芩苷在276 nm和318 nm波长处均有最大吸收,而木犀草苷在350 nm波长处有最大吸收,为了便于检测,选择350 nm为检测波长[2]。在此波长下检测,2种成分均有较大的响应,重复性好。
3.2 流动相的选择
试验中发现,选择甲醇-水作流动相时,混合压力明显增高;而选择乙腈-水作流动相时,乙腈的洗脱能力强,且黏度较小,柱压大小比较合适,但所得峰容易拖尾。后将乙腈-水换成乙腈-冰醋酸,所得的峰拖尾减少。再对冰醋酸的浓度进行调节(0.1%~1.0%),经过比较发现,文中所采用的色谱条件分离效果好,保留时间适宜。
3.3 提取方法的选择
试用2010年版《中国药典》(一部)HPLC法测定金银花中绿原酸和木犀草苷含量对样品的处理方法[3],加50%甲醇与70%乙醇稀释样品,超声20 min及采用中性氧化铝洗脱[3],分离效果均不理想。故选择用乙酸乙酯提取,残渣用50%甲醇溶解的提取方法[3]。该法制备的供试品溶液所得峰形、出峰时间及分离度均可达到较好效果。
[1]高益民,王忠山.对痰热清注射液临床药学初步评价[J].首都医药,2004,11(12):44.
[2]陈福超,李 鹏,方宝霞.HPLC法测定苦碟子注射液中木犀草素的含量[J].中国药师,2010,13(2):220.
[3]国家药典委员会编.中华人民共和国药典(一部)[S].2010年版.北京:中国医药科技出版社,2010:205-206、159、758-759.
