散结明目颗粒的制备及质量标准研究
2011-08-06张艳君马婷玉聂继红王萍刘兆龙新疆维吾尔自治区人民医院乌鲁木齐市83000新疆医科大学附属中医医院药学部乌鲁木齐市830000
张艳君,马婷玉,聂继红,王萍,刘兆龙(.新疆维吾尔自治区人民医院,乌鲁木齐市83000;.新疆医科大学附属中医医院药学部,乌鲁木齐市 830000)
散结明目方是由新疆医科大学附属中医医院眼科李全智主任医师创立的验方,临床长期用于治疗由于目络瘀阻引起的视瞻昏渺、暴盲,以及玻璃体积血和增殖性视网膜病变。该方具有活血祛瘀、散结明目等功效。为方便患者使用,本课题组拟将其制成颗粒剂。为全面控制制剂的内在质量,对方中夏枯草、赤芍、槐花、川芎等进行定性鉴别,并采用高效液相色谱(HPLC)法对处方中丹参的有效成分丹酚酸B进行含量测定[1]。
1 仪器与试药
2695HPLC仪(美国Waters公司);AL204电子天平、AG135电子天平(瑞士梅特勒-托利多仪器(上海)有限公司);SK3300LH超声清洗器(上海科导超声仪器有限公司);Di-rect-QTM5超纯水仪(美国Millipore公司)。
丹参、红花、赤芍、夏枯草、莪术、三棱、水蛭、槐花、茺蔚子、昆布、川芎药材均由新疆医科大学附属中医医院提供,经新疆医科大学附属中医医院副主任药师李玲检验均符合2010年版《中国药典》(一部)项下规定[2];丹酚酸B、熊果酸、芍药苷、芦丁对照品和夏枯草、赤芍、槐花、川芎对照药材(中国药品生物制品检定所,批号分别为110736-200630、110742-200516、110736-200630、 100080-200707、 120993-200604、 121093-200402、121270-200602、210918-200608);ZTC1+1-Ⅱ天然澄清剂,包括A、B组分(天津正天成澄清技术有限公司);甲醇、乙腈为色谱纯,水为超纯水,其他试剂均为分析纯。
2 处方与制备
2.1 处方
本品处方由丹参、夏枯草、赤芍、红花等11味中药组成。
2.2 制备
按处方比例称取夏枯草、赤芍等8味药材,加入10倍量80%乙醇提取3次,每次1 h;合并3次醇提液,将醇提液浓缩至药材与药液比为1∶10,缓慢细流加入药液量10%的ZTC+1-Ⅱ型天然澄清剂的B组分,边加边搅,置于60℃水浴中保温1 h,再缓慢细流加入药液量5%的ZTC+1-Ⅱ型天然澄清剂的A组分,搅匀,置于60℃水浴中保温1 h,静置24 h,离心2 min;浓缩干燥成干膏,干膏粉碎过80目筛,备用。
按处方比例称取丹参、红花、昆布3味药材,加入20倍量水提取2次,每次0.5 h[3,4];合并2次水提液,将水提液浓缩至药材与药液比为1∶4,4 000 r·min-1离心10 min;浓缩干燥成干膏,干膏粉碎过80目筛,备用。
取干膏粉量20%的莪术细粉(过80目筛)和5‰的甜菊糖苷与上述2种干膏粉混合均匀,用92%乙醇为黏合剂制粒,黏合剂加入量为40 mL·100 g-1。将制得颗粒进行干燥,干燥温度设定为40~60℃,干燥6 h,在干燥过程中,温度应逐渐升高。将干燥后聚结成块的颗粒过14目筛整粒,即可。
3 定性鉴别
3.1 夏枯草的薄层色谱(TLC)鉴别
取3批样品的细粉各1 g,加乙醇20 mL,加热回流1 h,滤过,滤液蒸干,用石油醚(30~60℃)浸泡2次,每次15 mL(约2 min),倾去石油醚液,残渣加乙醇1 mL使溶解,作为供试品溶液;取夏枯草对照药材1 g,同法制成对照药材溶液;另取熊果酸对照品,加乙醇制成每1 mL含0.2 mg的溶液,作为对照品溶液;取处方中除夏枯草以外的所有药材,按制备工艺制得阴性样品,照供试品溶液制备方法制成阴性对照溶液。照TLC法[2]试验,吸取上述4种溶液各10 μL,分别点于同一硅胶G薄层板上,以环己烷-三氯甲烷-乙酸乙酯-冰醋酸(20∶5∶8∶0.5)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,于100℃加热至斑点显色清晰,分别置于日光、紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照品、对照药材色谱相应的位置上,分别显相同颜色的斑点或荧光斑点;阴性对照无干扰。夏枯草的TLC分别见图1、图2。
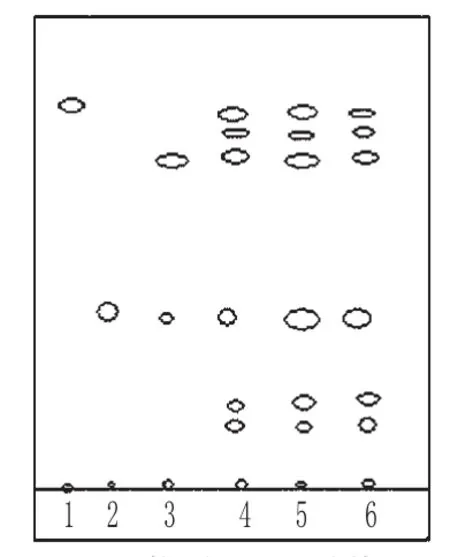
图1 夏枯草的TLC(紫外光)Fig 1 TLC of Prunella vulgaris(ultraviolet light)

图2 夏枯草的TLC(日光)Fig 2 TLC of P.vulgaris(sunlight)
3.2 赤芍的TLC鉴别
取3批样品各0.5 g,加乙醇10 mL,振摇5 min,滤过,滤液蒸干,残渣加乙醇2 mL使溶解,作为供试品溶液;取赤芍对照药材0.5 g,同法制成对照药材溶液;取处方中除赤芍以外的所有药材,按制备工艺制得阴性样品,同法制成阴性对照溶液;另取芍药苷对照品,加乙醇制成每1 mL含2 mg的溶液,作为对照品溶液。照TLC法[2]试验,吸取上述4种溶液各10 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(40∶5∶10∶0.2)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,加热至斑点显色清晰。结果,供试品色谱中,在与对照品、对照药材色谱相应的位置上,分别显相同颜色的斑点或荧光斑点;阴性对照无干扰。赤芍的TLC见图3。
3.3 槐花的TLC鉴别
取3批样品的细粉各0.2 g,加甲醇5 mL,密塞,振摇10 min,滤过,滤液作为供试品溶液;取槐花对照药材0.5 g,同法制成对照药材溶液;取处方中除槐花以外的所有药材,按制备工艺制得阴性对照样品,同法制成阴性对照溶液;另取芦丁对照品,加甲醇制成每1 mL含0.5 mg的溶液,作为对照品溶液。照TLC法[2]试验,吸取上述4种溶液各15 μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-甲酸-水(8∶1∶1)为展开剂,展开,取出,晾干,喷以三氯化铝试液,待乙醇挥干后,分别置于日光、紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照品、对照药材色谱相应的位置上,分别显相同颜色的斑点或荧光斑点;阴性对照无干扰。槐花的TLC分别见图4、图5。
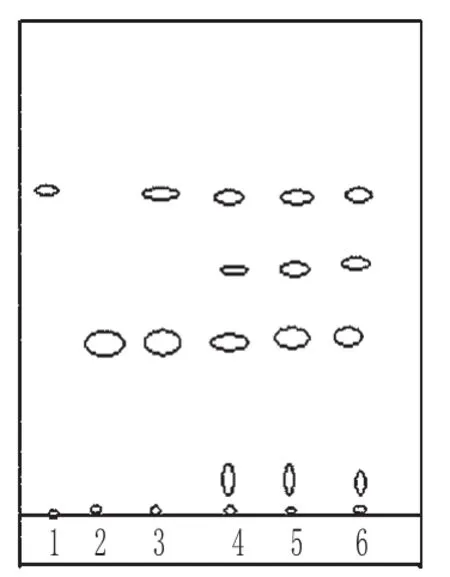
图3 赤芍的TLCFig 3 TLC of Paeoniae Radix Rubra

图4 槐花的TLC(紫外光)Fig 4 TLC of Sophora japonica(ultraviolet light)
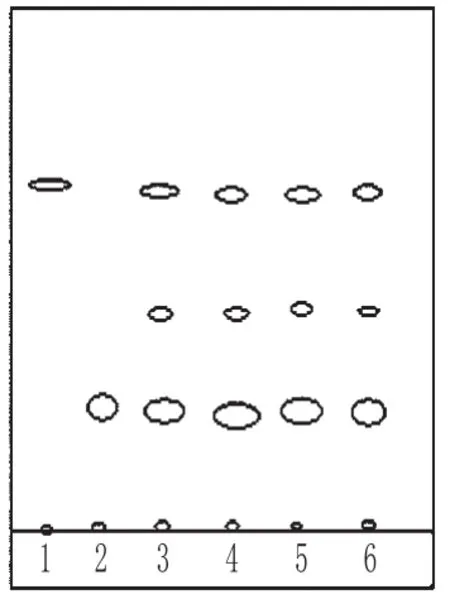
图5 槐花的TLC(日光)Fig 5 TLC of S.japonica(sunlight)
3.4 川芎的TLC鉴别
取3批样品的细粉各2.9 g,置于锥形瓶中,加入20 mL乙醚,密塞,振摇称重后回流1 h,过滤,将滤液蒸干,残渣加入乙酸乙酯20 mL使溶解,作为供试品溶液;取川芎对照药材1 g,同法制成对照药材溶液;取处方中除川芎以外的所有药材,按制备工艺制得阴性样品,同法制成阴性对照溶液。照TLC法[2]试验,分别吸取上述3种溶液各15 μL,点于同一硅胶G薄层板上,以正己烷-乙酸乙酯(9∶1)为展开剂,展开,取出,晾干,在紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点;阴性对照无干扰。川芎的TLC见图6。
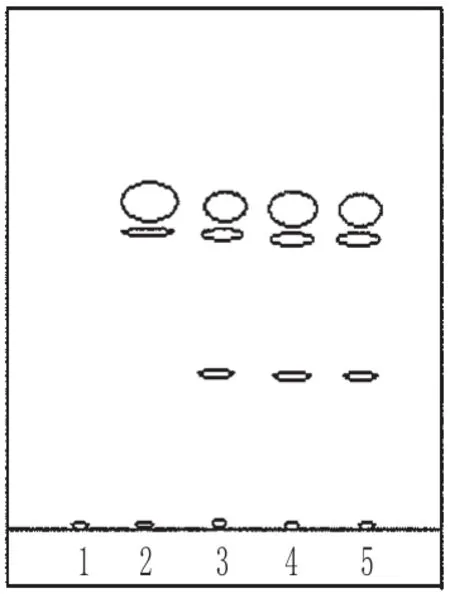
图6 川芎的TLCFig 6 TLC of Ligusticum chuanxiong
4 含量测定
4.1 色谱条件
色谱柱:SHIMADZU VP-ODS(150 mm×4.6 mm,5 μm);流动相:乙腈-0.2%磷酸(23.5∶76.5);检测波长:286 nm;柱温:30 ℃;流速:1.0 mL·min-1;进样量:10 μL。
4.2 方法学考察
4.2.1 供试品溶液的制备 精密称取研至细粉的样品颗粒2 g,置于锥形瓶中,加入80%甲醇20 mL,密塞,振摇,称重,超声20 min,补足失重,滤纸滤过,即得。
4.2.2 对照品贮备液的制备 精密称取丹酚酸B对照品38.75 mg,加甲醇适量溶解,并定容至25 mL量瓶中,作为对照品贮备液。
4.2.3 阴性对照试验 按处方量精密称取除丹参外其余10味药材,按制备工艺制成阴性样品,精密称取研至细粉的阴性样品2 g,照“4.2.1”项下方法制备成缺丹参的阴性对照溶液。在上述色谱条件下测定,结果阴性对照干扰。色谱见图7。

图7 高效液相色谱图Fig 7 HPLC chromatograms
4.2.4 线性关系考察 分别吸取对照品贮备液0.2、1.0、1.8、2.6、3.4 mL,置于5 mL容量瓶中,用甲醇定容,摇匀浓度分别为0.062、0.310、0.558、0.806、1.054 mg·mL-1的对照品系列溶液,微孔滤膜(0.45 μm)过滤后取10 μL注入色谱仪,照上述色谱条件测定。以峰面积积分值(Y)为纵坐标,检测浓度(X)为横坐标,进行线性回归,得回归方程为Y=7.763 2×10-8X+1.938 8×10-3(r=0.999 9)。结果,丹酚酸B检测浓度在0.062~1.054 mg·mL-1范围内与峰面积积分值呈良好的线性关系。
4.2.5 精密度试验 取同一对照品溶液,在上述色谱条件下连续进样5次,记录丹酚酸B的峰面积。结果,RSD=1.61%(n=5),表明仪器精密度良好。
4.2.6 加样回收率试验 取已知含量的供试品溶液(含量:0.690 1 mg·mL-1)9份,按供试品溶液中含量的80%、100%、120%加入不同量的丹酚酸B对照品溶液,制备成高、中、低浓度的各3份样品,按“4.2.1”项下方法制备供试品溶液,在上述色谱条件下测定,计算加样回收率。结果,平均回收率为100.3%,RSD=1.52%(n=9)。
5 讨论
本试验对处方中的所有药味分别进行了TLC鉴别。结果,夏枯草、赤芍、槐花、川芎的TLC鉴别斑点清晰,分离度好,阴性对照无干扰,因此列入质量标准。
由于颗粒提取成分复杂,薄层层析干扰较大,在其他成分的薄层展开过程中反复出现以下问题:对照品斑点与供试品斑点比移值有差别,阴性有干扰;目标斑点与其他组分斑点分离度不够,造成斑点叠加。处方中其他成分的TLC鉴别有待进一步研究。
研究制剂中丹酚酸B含量测定方法时,曾选用水、甲醇、乙醇等不同的溶剂进行提取,并采用加热回流提取和超声提取两种不同的方法进行提取。结果,甲醇超声提取简便、快捷,且提取率较高,因此选用甲醇作为提取溶剂。笔者曾比较了不同比例的甲醇-乙腈-2%甲酸、甲醇-乙腈-1%甲酸、乙腈-0.2%磷酸等多种溶剂系统作为流动相,结果以乙腈-0.2%磷酸(23.5∶76.5)作为流动相,色谱图峰形好,出峰时间适宜,分离效果较好。
[1] 杨 男,胡 明,蒋学华.国外临床药学服务的质量标准及其对我国的启示[J].中国药房,2009,20(7):486.
[2] 国家药典委员会编.中华人民共和国药典(一部)[S].2010年版.北京:中国医药科技出版社,2010:70、141、147、263、257、12、77、333、225、195、38、附录34.
[3] 于沈晶,殷文广,修志龙.丹参不同提取工艺的研究[J].中草药,2007,38(4):542.
[4] 聂继红,周 玲.复方中丹参提取工艺及含量测定的研究[J].中成药,2007,29(6):889.
