胚胎植入前遗传学检测在阻断常染色体隐性多囊肾病家系多囊肾/多囊肝病变1基因新突变遗传的应用
2024-04-15王凝郝燕陈大蔚章志国匡丹张清尹奕琪魏兆莲周平曹云霞
王凝 郝燕 陈大蔚 章志国 匡丹 张清尹奕琪 魏兆莲 周平 曹云霞
1安徽医科大学第一附属医院妇产科 (合肥 230000);2国家卫生健康委配子及生殖道异常研究重点实验室(合肥 230000);3出生人口健康教育部重点实验室 (合肥 230000);4生殖健康与遗传安徽省重点实验室(合肥 230000);5安徽省生命资源保存与人工器官工程技术研究中心 (合肥 230000); 6安徽省转化医学研究院 (合肥 230000)
常染色体隐性遗传性多囊肾病(autosomal recessive polycystic kidney disease, ARPKD)为单基因常染色体隐形遗传病,致病基因是多囊肾/多囊肝病变1 基因(polycystic kidney and hepatic disease 1, PKHD1)或DAZ 相互作用蛋白1 样基因(DAZ interacting zinc finger protein 1 like, DZIP1L)[1-2]。ARPKD 的发病率约为1∶10 000~1∶40 000,比常染色体显性多囊肾病(ADPKD)更罕见且更严重。ARPKD 发病早且十分严重,30%~50%患儿于围产期或< 1 岁时即因肺功能发育不全发病死亡。存活的患儿肾功能将进行性恶化,最终进展至终末期肾病(ESRD),预后很差,成年患者罕见[1,3]。ARPKD 尚无治愈性干预措施,其治疗为对症支持治疗(包括长期肾替代治疗),患者并发症多、生存质量差、治疗费用昂贵[4]。胚胎植入前遗传学诊断(PGD)一直用于筛查患有已知遗传疾病的夫妇的正常胚胎,以避免通过产前诊断终止受影响胎儿的妊娠。对于PKHD1 基因突变携带的父母,可通过植入前遗传学检测(preimplantation genetic testing, PGT)筛选正常胚胎,或产前诊断筛查患病胎儿来预防ARPKD患儿的出生。常规产前诊断只能在妊娠后进行,一旦确诊即需治疗性引产。而PGT可以在胚胎植入母体子宫前对胚胎进行检测,是一种更早期的产前诊断方法,可以避免妊娠后的选择性流产对孕妇产生的身心创伤。ARPKD 广泛的表型和遗传异质性使得需要考虑的基因数量不断增加。相较于传统的通过PCR 直接检测突变或短串联重复序列(short tandem repeat, STR)连锁分析,基于二代测序(next generation sequencing, NGS)的单核苷酸多态性(single nucleotide polymorphism, SNP)连锁分析具有更高的检测效率、更低的基因脱扣率、更确定的基因型-表型相关性,可以帮助克服PKHD1 基因的复杂性和突变位点的不确定性,实现定位克隆和产前诊断。国内外运用基于NGS 技术的SNP 连锁方法对ARPKD家系进行PGT 的报道较少,本研究在明确PKHD1基因致病突变的情况下,应用NGS 对胚胎行SNP连锁分析,活产一健康婴儿,阻断单基因病在家系中的垂直传递,为患者家庭的优生优育提供保障。
1 资料与方法
1.1 观察对象先证者父亲(Ⅰ1):26 岁,PKHD1基因c.10444C > T(p.R3482C),表型正常,既往体健。先证者母亲(Ⅰ2):26 岁,PKHD1 基因c.4303del(p.S1435Vfs*25)(图1),表型正常,既往体健,G1P0A1,多囊肾胎儿(先证者Ⅱ1)引产史,携带父母双方PKHD1 基因突变,其中c.4303del突变为首次报道突变。为了避免再次妊娠患儿,这对夫妇于2021 年8 月在安徽医科大学第一附属医院生殖医学中心经过遗传咨询后,要求接受试管婴儿+ 单基因病检测(PGT for monogenic gene defects, PGT-M)+ 非整倍体检测(PGT for aneuploidies, PGT-A)。已向这对夫妇充分解释PGT过程及相关风险,获得其完全知情同意。本研究经安徽医科大学第一附属医院生殖医学伦理委员会批准(编号:2017002)。
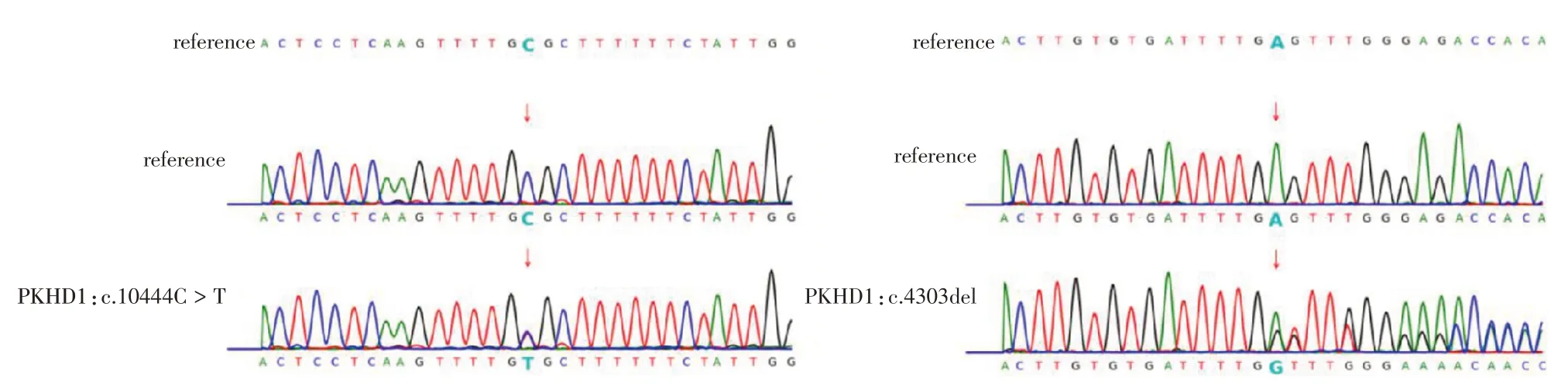
图1 先证者PKHD1 基因c.10444C > T(父源)和c.4303del(母源)突变Sanger 测序图Fig.1 Sanger sequencing diagram of c.10444C > T (paternal origin) and c.4303del (maternal origin) mutations in the proband's PKHD1 gene
1.2 方法
1.2.1 构建先证者和父母双方SNP 单体型提取引产胎儿皮肤组织和男女双方外周血DNA,在突变位点两侧2M 区域内选择若干信息丰富、紧密连锁的SNP,提交给Ion AmpliSeq Designer 网站进行引物设计。经多重置换扩增(multiple displacement amplification, MDA)、多重PCR、纯化、建库、富集后,采用NGS 测序、选择有效SNPs 来建立SNP 单倍型。
1.2.2 胚胎培养、活检及囊胚玻璃化冷冻黄体期长方案促排卵后,采用卵胞浆内单精子注射方式进行授精,受精后继续培养至第5 天或第6 天,待囊胚形成后在激光辅助下取大约5~10 个滋养外胚层(TE)细胞进行活检。活检的囊胚单管单枚玻璃化冷冻处理,编号。
1.2.3 PGT-M 与PGT-A 联合诊断对活检细胞进行多重置换扩增(multiple displacement amplification, MDA)、多重PCR、纯化、建库、富集后,采用Sanger 测序直接检测PKHD1 基因突变和基于NGS的SNP 连锁分析鉴别携带突变的染色体两种方法进行PGT-M,同时进行拷贝数变异(copy number variation, CNV)分析以进行低深度的PGT-A。
1.2.4 FET 和随访评估使用自然周期来准备子宫内膜进行胚胎解冻移植(frozen-thawed embryo transfer, FET)。生化妊娠在移植后14 d 查血人绒毛膜促性腺激素(hCG)确定;移植后4 周行经阴道B 超查看孕囊确定宫内妊娠,移植后6~8 周经阴道B 超查看胎心排除胎停,孕18~24 周时行羊膜腔穿刺术获得胎儿DNA 行Sanger 测序验证PGT结果。
2 结果
2.1 先证者父母和先证者的SNP 单倍型选择突变位点两侧2M 区域内的335 个信息丰富的SNP成功构建先证者及其父母的SNP 单倍型,区别确认了父母的正常及风险染色体。
2.2 体外受精结果获卵数20 枚,MII 数19 枚,受精数17 枚,卵裂17 枚,其中双原核受精数16 枚,2PN 卵裂数16 枚。
2.3 植入前遗传学检测将培养第5 天后第6 天得到的优质囊胚分别标记为D501、D601、D602、D603、D604 和D605,行TE 活检。PGT 结果显示,6 个胚胎中,D601、D603、D604、D605 未检测到突变且为整倍体胚胎,D501 携带PKHD1 基因c.4303del突变(杂合子)且6 号染色体三体嵌合,D602 测序数据波动大,无法判断。
2.4 FET 结果选择1 枚未检测到突变的优质整倍体胚胎进行冻融胚胎移植(D601),在第3 个月经周期行单囊胚解冻移植。移植14 d 后外周血hCG 阳性,移植40 d 后经阴道超声见单个孕囊,大小为43 mm × 40 mm,胚芽长21.9 mm,可见心管搏动。孕18 周经羊膜腔穿刺获得胎儿DNA,Sanger 测序未检测到PKHD1 基因c.10444C > T 和c.4303del 突变,证实了PGT 结果。孕38+2周顺产一正常男婴,体质量3.05 kg,健康状况良好。
3 讨论
临床较常见的遗传性肾病是ADPKD,患病率约1/1 000,主要由PKD1 或者PKD2 基因突变引起[5]。而本研究中为常染色体隐性多囊肾病,是由PKHD1基因突变或DZIP1L 基因突变导致的常染色体隐形遗传病,发病率仅为1∶10 000~1∶40 000,要罕见的多。致病基因PKHD1 位于第6 号染色体6p21.1-p12,长约472 kb,编码蛋白为4 047 个氨基酸的纤囊素(fibrocystin/polyductin, FPC)。FPC 缺陷可引起纤毛功能紊乱、肾小球囊内上皮细胞异常增殖和转运异常,最终导致肾小管、胆管囊性扩张及纤维化[6-7]。
ARPKD 大多发病早且严重,胎儿期即可出现双侧肾脏增大、皮髓质分化差、微小囊肿形成、肺功能发育不全,可导致30%~50%的新生儿死亡。新生儿期存活的患儿可相继出现电解质紊乱、代谢性酸中毒及高血压等肾小管功能异常的症状,并随年龄增长进行性恶化,最终进展至ESRD,并伴有肝纤维化进行性加重,导致门脉高压,预后较差,成年患者罕见[3-10]。
PKHD1 具有复杂的基因型-表型相关性,其大尺寸、复杂的剪接模式和转录谱、广泛的等位基因异质性、大量错义突变和新突变以及我们对蛋白质功能的有限理解为建立PKHD1 的基因型-表型相关性带来了挑战。鉴于这些挑战,基因型-表型相关性侧重于突变的类型,而不是突变的特定位点[11]。几乎所有携带两个截断突变的患者都显示出严重的表型和新生儿期死亡。在新生儿期存活的患者通常至少有一个错义突变,但是反过来,一些错义变化可能和截断突变一样具有破坏性[12]。
PKHD1 基因以编码区的点突变致病为主[13],目前,HGMD(www.hgmd.cf.ac.uk)中报道的PKHD1突变种类已达911 种,其中约65.75%为错义/无义突变,15.26%为小片段缺失,4.10%为小片段插入,1.20%为小片段插入缺失,3.62%为大片段缺失,0.04%为大片段插入,0.02%为复合突变,其中c.107C > T(p.Thr36Met)是最常见的突变。本研究中先证者携带的PKHD1 基因c.4303del(p.S1435Vfs*25)突变为首次报道,该变异为截断突变,根据美国医学遗传学与基因组学学会(ACMG)指南,提示为致病突变位点,该突变的发现丰富了PKHD1基因的突变数据库,针对该突变行PGT,亦为首次。
7 例由纤毛相关基因DZIP1L 突变导致的ARPKD患者在2017年由LU等在4个无血缘关系的家族中首次发现。DZIP1L 位于3 号染色体(3q22.1-q23),具有16 个外显子,编码767 个氨基酸的蛋白质,定位于纤毛过渡区,负责将基因产物转运到睫状轴突[2]。与PKHD1 突变相比,DZIP1L 突变是ARPKD 的罕见原因,且迄今为止描述的DZIP1L 突变患者的临床病程总是中等的,没有一个显示围产期死亡。
正常人群中PKHD1 突变杂合子携带率约为1/70,携带者夫妻每次妊娠的孩子有25%的风险患病,50%的风险携带致病基因。胎儿肾脏异常在妊娠中晚期才能通过超声检查发现[14-16]。对于ARPKD 高风险家庭,基因诊断是金标准。常规产前诊断方法通常在孕中期行羊水穿刺进行诊断,若发现胎儿为ARPKD,则孕妇会面临治疗性引产带来的身心伤害。而PGT 在胚胎植入前进行遗传学检测,是一种更早期的产前诊断方法,可以避免因妊娠ARPKD 胚胎或非整倍体胚胎而导致的流产,可以阻断PKHD1 突变基因在该家系中的垂直传递。因此,PGT 与传统的产前诊断相比具有明显的优势[13,16]。
等位基因脱扣(ADO)是可能导致PGT-M 误诊、可移植胚胎数目减少的重要问题[17]。ADO 是由等位基因不等扩增引起的某些等位基因未检测到,是单细胞PCR 的固有缺陷。PKHD1 较大且ARPKD 相关变异数量众多,直接测序漏诊率很高,需要更灵敏的检测方法降低ADO 率[18]。SNPs指在基因组水平上由单个核苷酸的变异所引起的DNA 序列多样性,在群体中的频率> 1%。SNP 连锁分析方法间接诊断胚胎基因型方法如下:通过NGS 或芯片检测父母和胚胎的目标基因上下游1-2M 内SNPs 作为遗传标记,在家系中构建单倍型,再通过检测胚胎是否携带风险单体型来判断胚胎是否携带基因突变。由于采用SNP 作为遗传标记构建了单体型,对胚胎进行基因检测时,可检测位点大大增加,且NGS 具有高通量、高覆盖率和高灵敏度的特征,所以相比于传统的通过PCR 方法直接检测突变或STR 连锁分析,基于NGS 的SNP 连锁分析具有更高的检测效率、更低的等位基因脱扣率、更确定的基因型-表型相关性[19-21]。作为遗传标记,SNP 与STR 相比,有以下优点:(1)SNP 是二态性的,只需+/-的分析,适于快速、规模化筛查;(2)SNP 数量更大、分布更广泛;(3)SNP更稳定;(4)部分SNP 本身可能就是疾病遗传机制的候选改变位点;(5)与传统的凝胶电泳相比,SNP 芯片具有廉价、快速的优点;(6)SNP 连锁分析可以直接对突变位点进行检测,实现基因型和单体型的同时诊断[22-23]。本研究中通过连锁分析方法,在突变位点两侧2M 区域内选择335 个信息丰富、紧密连锁的SNP 位点作为遗传连锁标记,成功构建先证者父母的单体型,说明该方法是安全有效的,减少了因等位基因脱扣导致的误诊[13]。有1 枚胚胎未获明确诊断,主要考虑是活检细胞数目偏少,导致扩增失败。
SNP 连锁分析的局限性在于需要完整家系来构建家系单体型来进行诊断,在无法获取先证者DNA 的情况下,SNP 单体型方法无法奏效。在发生减数分裂重组的情况下,SNP 单体型分析也有可能存在无法诊断的情况。若患者卵巢功能较差,无法获取足够卵子和胚胎,患者行PGT 的成功率会大大降低。本研究中,无上述情况,可进行常规SNP 连锁分析。在一些特殊情况下,如没有患儿DNA 保存,可尝试通过致病胚胎极体或单精子构建家系单体型[24]。也可以通过三代长读测序,直接寻找患者SNP 位点,而不依赖于家系成员[25]。
综上所述,本研究应用基于NGS 的SNP 连锁分析技术,成功对ARPKD 家系进行了准确、高效的PGT-M,降低了等位基因脱扣导致的PGT-M 误诊率,避免了因选择患病或非整倍体胚胎而导致的流产问题,阻断了ARPKD 在该家系中的垂直传递,为未来预防ARPKD 出生缺陷的提供了参考,并且发现了PKHD1 基因的一个新突变,丰富了ARPKD 的突变数据库。
【Author contributions】WANG Ning performed the experiments and wrote the manuscript.HAO Yan performed the experiments and helped to revise the manuscript.CHEN Dawei, ZHANG Zhiguo,KUANG Dan, ZHANG Qing and YING Yiqi helped to recruit the patients.WEI Zhaolian, ZHOU Ping and CAO Yunxia analysed the data.All authors read and approved the final manuscript as submitted.
【Conflict of interest】The authors declare no conflict of interest.
