SCN4A基因R669H变异的家族性低钾性周期性麻痹1家系分析
2024-03-11李洁王乐黄雪霜姜海鸥
李洁,王乐,黄雪霜,姜海鸥
低钾性周期性麻痹(HOKPP)于1863年由Cavare首先报道,发病率约为1/100000[1]。该病是一种罕见的离子通道疾病,呈现不规则显性遗传模式,其外显率存在男女性别差异,国内男性高于女性,国外二者基本相同[2]。HOKPP的主要临床特点表现为周期性的骨骼肌弛缓性瘫痪,发作时伴有血钾水平降低,血清肌酶升高[3]。目前,研究[4-5]发现SCN4A、CACNAIS和KCNE3基因是HOKPP的致病基因,分别编码人骨骼肌Na+通道、Ca2+通道和K+通道蛋白。该病具有高度的临床和遗传异质性,即使相同的突变在不同种族或同一家系不同患者之间也会导致临床表型差异。本研究报道一个HOKPP家系,并采用全外显子组测序(WES)和Sanger测序技术进行突变分析[6],旨在明确此家系的致病基因变异位点和临床表型之间的关系。
1 临床资料
1.1 先证者 先证者,苗族,男,22岁,因“四肢无力9 h”入院。病情初发时尚可行走,约1 h后出现双下肢软瘫,伴有肌痛,头晕。直至目前,反复发作四肢无力14年,凌晨发作较多,发作诱因主要为酗酒和剧烈运动,每次均血钾低(1~2 mmol/L),补钾后恢复。近年来,发作频次增多,每年发作3~5次,症状加重,出现双下肢软瘫,伴有肌痛。既往有“低钾血症”病史,无甲状腺功能亢进及高血压病史。查体:双下肢肌力Ⅱ级,肌张力正常,余未见明显异常。实验室检查:血钾1.48 mmol/L,血钠、血镁和血钙正常。血气分析、血生化、尿常规、肾功能及甲状腺功能检查均未见明显异常。采用WES方法和Sanger测序验证在SCN4A基因上发现1个杂合错义突变c.2006G>A,该变异导致cDNA的第2006位碱基G被A替换(图1),使其编码的蛋白质多肽链中第669位精氨酸被组氨酸所取代(p.R669H),经美国医学遗传学和基因组学学会(ACMG)指南评估此变异为致病性变异。诊断:HOKPP。入院后给予补钾,患者症状好转。发作间期血钾正常。
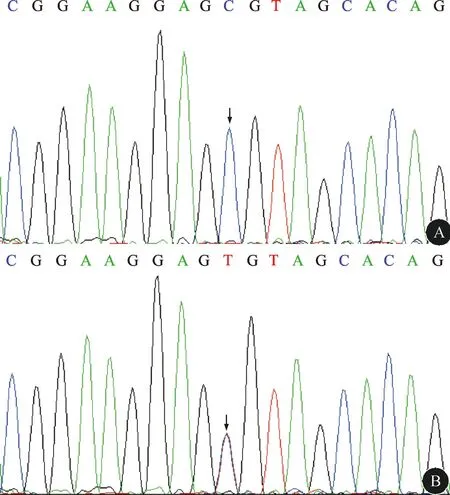
图1 SCN4A基因反向测序的结果A:先证者、先证者表哥、先证者父亲及先证者小姑为c.2006C>T变异;B:先证者大姑及叔叔为野生型。
1.2 家系分析 家系图见图2。该家系4代共有5人患病,均为男性,连续遗传3代,呈现常染色体显性遗传模式。先证者发病年龄为8岁,表哥为10岁,父亲为14岁,爷爷不详(已故)。先证者父亲经自身严格管理,避免诱发因素,起病至今仅发作2次;先证者表哥发作频率为3~5次/年;先证者爷爷为2~3次/年。该家系发病年龄有提前的趋势,发病频次明显增加。先证者表哥与先证者一样,凌晨发作较多,表现为四肢无力,周期性瘫痪,发作时查血钾极低(1~2 mmol/L),血气分析、血生化、尿常规、肾功能及甲状腺功能等检查均未见异常。诊断:遗传性HOKPP。PCR-Sanger测序验证结果显示,SCN4A基因上发现1个杂合错义突变c.2006G>A存在于先证父亲、小姑和表哥中,而其大姑及叔叔未发现该变异,其小姑尚无HOKPP临床表现。
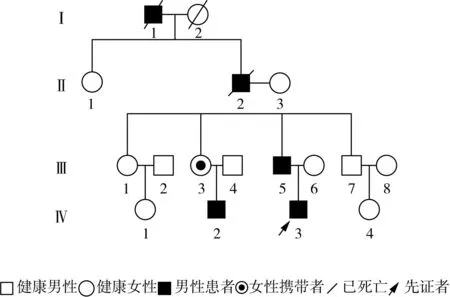
图2 HOKPP家系系谱图
2 讨 论
HOKPP是一种罕见的遗传性神经肌肉病,呈现散发性和家族性。与欧洲国家相比,亚洲各国以散发性为主[7],阳性家族史很少。家族性HOKPP通常表现为不规则显性遗传,其外显率因致病基因突变位点而不同,国外报道男性外显率与女性基本相同,而国内报道男性高于女性[2]。本研究报道HOKPP一家系,有明确的阳性家族史,家族中无隔代遗传现象,连续三代均有发病,其遗传方式为常染色体显性遗传。此家系与国内报道一致,发病者均为男性,但根据系谱图推测先证者小姑应为致病突变携带者,基因检测结果也得到了证实,可见该家系符合不规则显性遗传,存在外显不全。
HOKPP患者在诱发因素、起病年龄、电生理、发作频次及病理表型等方面具有高度异质性,但典型临床症状基本相似,发病时均表现为周期性骨骼肌弛缓性瘫痪,伴有明显血钾降低,临床通过补钾可有效治疗[8]。HOKPP严重时可导致肌无力、气促、胸闷,甚至肌肉变性[9],最终患者可死于低钾造成的恶性心律失常或呼吸肌麻痹。该家系患者在饮酒和剧烈运动之后,出现不同程度的血钾降低,双下肢软瘫,而且发病年龄有提前的趋势,发病频次明显增加,表现为高度的临床异质性。预防HOKPP发生的主要措施包括尽量防止诱发因素,胰岛素、肾上腺素、糖皮质激素等药物慎用;避免低钠和低碳水化合物及低钾饮食;对于频繁发作者可长期服用氯化钾等药物进行预防[10]。本家系中先证者父亲日常生活中通过避免诱发因素,至今发作2次,提示HOKPP进行积极管理可有效预防发作。
目前发现的与HOKPP相关的已知基因主要包括CACNA1S、SCN4A和KCNE3,分别编码人骨骼肌的Ca2+通道、Na+通道和K+通道蛋白。在美国和欧洲国家,导致HOKPP的主要原因是CACNA1S基因突变[11]。在中国,导致HOKPP最常见的原因是SCN4A基因突变,其次是KCNE3突变,CACNA1S突变发生率相对较低[11-12]。本家系通过WES和Sanger测序检测到3例男性患者和1位正常女性存在SCN4A基因杂合错义突变c.2006G>A,导致该基因编码的蛋白质多肽链第669位精氨酸被组氨酸所取代(p.R669H),根据ACMG指南评估该变异具有明确的致病性。据报道[13],R669H变异虽然对Na+通道的激活或快速失活无显著影响,但对慢速失活有增强作用,从而使Na+通道的连续电流增加2~5倍,导致患者Na+通道的功能异常,不能引起骨骼肌兴奋-收缩偶联并产生肌无力。该变异的主要临床特征[14]:发作时四肢反射消失;发病后出现肌痛;血钾水平偏低;补钾治疗有效,乙酰唑胺治疗却不能改善患者临床症状,甚至加重,这可能与SCN4A基因不同突变位点相关[10,15]。该家系患者与以上临床表现基本类似,但国外研究[16]表明家系中男女性外显率均为100%,而该家系的基因测序发现男性携带者完全外显,但女性携带者(Ⅲ3)未发病。我国HOKPP家系已报道过SCN4A基因R669H变异,但未明确女性是否为携带者[8]。本研究首次在中国HOKPP家系中发现了R669H变异在女性中可存在临床表型不外显。
总之,本研究不但为该家系的疾病诊断和遗传咨询提供了理论依据,而且证实在亚洲种族人群中SCN4A基因突变可造成女性为携带者,而男性完全外显,进一步丰富了HOKPP的临床表型和基因型之间的关系资料库。
作者贡献说明李洁完成实验操作、采集、统计分析数据;王乐指导文章修改,提供技术材料;黄雪霜指导选题、设计研究,提供研究经费;姜海鸥提出选题,设计、实施研究,提供研究经费,撰写、修改文章
