改良QuEChERS技术结合超高效液相色谱-串联质谱法测定水产品中扑草净及其代谢物残留
2024-03-10穆迎春喻亚丽陈建武甘金华
彭 婕,穆迎春,喻亚丽,陈建武,刘 婷,何 力,3,*,甘金华,3,*
(1.中国水产科学研究院长江水产研究所,农业农村部淡水鱼类种质监督检验测试中心,农业农村部水产品质量安全风险评估实验室(武汉),湖北 武汉 430223;2.中国水产科学研究院,北京 100141;3.农业农村部水产品质量安全控制重点实验室,北京 100141)
扑草净是一种选择内吸传导型均三氮苯类除草剂,在中国农业生产和水产养殖中应用广泛[1],它具有类似苯环的结构,化学性质比较稳定,是一种持久性有机污染物[2]。20世纪70年代,扑草净开始被用于池塘水产养殖中清除杂草和青苔[3],后续又用于去除虾类、海参、贝类等养殖中的大型藻类[4]。种植业和渔业使用的扑草净大部分随着地表径流和雨水冲刷等途径进入水体环境[5],它在水中有较高的溶解度,难以降解,经过自然界的水循环作用被带入江河湖海,从而导致国内外不同水域中均能检测到扑草净残留[6-10]。扑草净通过水体环境在生物体内富集,沿食物链传递,危害人类健康[11],它会引起生物机体的内分泌紊乱、肿瘤产生等不良危害[12-13],因此,美国和欧盟等已将其列入内分泌干扰物名单[14],中国也于2010年在中华人民共和国农业部公告第1435号中将其列入《兽药试行标准废止目录》[15]。近年来我国水产品中多次检出扑草净残留[16-17],甚至突发使用扑草净清除虾池中的青苔,导致克氏原螯虾全部死亡的事故,2021年扑草净被正式纳入“国家产地水产品兽药残留监控计划”主要检测指标,可见水产品中扑草净残留问题已引起广泛的关注和重视。扑草净在动物体内经降解代谢转化途径产生了结构多样的代谢中间产物[18],其中主要包含通过N-脱烷基化反应形成的化合物去异丙基扑草净(desisopropyl prometryn,DIP)和双去异丙基扑草净(dideisopropyl prometryn,DDIP),硫氧化形成的化合物扑草净亚砜,以及羟基取代甲硫基形成的化合物2-羟基-扑灭津等[19]。目前诸多研究都只针对水产品中扑草净残留分析,缺乏有效的水产品中扑草净及其代谢产物残留测定方法,无法准确评估水产品中扑草净残留风险,因此有必要开发水产品中扑草净及其主要代谢产物DIP、DDIP、扑草净亚砜和2-羟基-扑灭津快速准确的分析方法,进一步提高水产品质量安全监测水平。
由于水产品基质具有组分复杂、干扰物质多以及目标物含量低等特点,因此,准确评估水产品中扑草净及其代谢物的残留水平需要优良的前处理方法和精准的分析技术。目前报道的扑草净残留测定方法主要有气相色谱法[1]、气相色谱-质谱联用法[20-21]、气相色谱-串联质谱法[22]和液相色谱-串联质谱法[23-24]。对比4 类分析方法,气相色谱法灵敏度不高,准确定性能力较弱;气相色谱-质谱联用法和气相色谱-串联质谱法在精准定性能力方面有所提升,但检测灵敏度仍不具备显著优势;液相色谱-串联质谱法由于其灵敏度高、选择性好,成为分析复杂基质中扑草净残留的首选方法。当前,水产品中扑草净残留分析多采用固相萃取(solid phase extraction,SPE)净化[25-27],此类方法往往需先活化SPE柱,再上样、淋洗、洗脱,不仅过程耗时,还需消耗大量溶剂,既不经济也不环保。近年来,QuEChERS(quick,easy,cheap,effective,rugged and safe)作为一种快速、简单、廉价、高效、安全的前处理技术,在动物性食品农兽药残留检测中应用较为广泛[28-30]。
本研究经实验优化,采用增强型脂质去除吸附剂(enhanced matrix removal of lipids,EMR-Lipid)与Cleanert LipoNo混合物作为净化吸附剂,基于改良的QuEChERS技术结合超高效液相色谱-串联质谱分析、同位素内标定量,建立了一种同时测定水产品中扑草净及其代谢物残留的分析方法。该方法操作简便、定量结果准确、灵敏度高,具有良好的准确度和精密度,以期为水产品中新型持久性有机污染物残留来源和质量安全评价提供技术支撑。
1 材料与方法
1.1 材料与试剂
草鱼、克氏原螯虾、中华绒螯蟹样品来源于本地农贸市场。
扑草净标准品(CAS 号7287-19-6、分子式C10H19N5S,纯度≥99%)、DDIP标准品(CAS号5397-01-3、分子式C4H7N5S,纯度为99.7%)、2-羟基-扑灭津标准品(CAS号7374-53-0、分子式C9H17N5O,纯度为99.9%)天津阿尔塔科技有限公司;DIP标准品(CAS号4147-57-3、分子式C7H13N5S,纯度≥99%)美国TLC公司;扑草净亚砜标准品(CAS号55702-48-2、分子式C10H19N5OS,纯度≥99%)美国Panphy公司;扑草净-D6标准品(CAS号1705649-52-0、分子式C10H13D6N5S,纯度≥98%)美国Dr.Ehrenstorfer公司;扑草净原药(纯度>97%)浙江中山化工集团有限公司。
乙腈、乙酸乙酯(均为色谱纯)美国J.T.Baker公司;甲醇、乙腈(均为质谱级)德国Meker公司;甲酸(质谱级)美国Fisher公司;氯化钠、无水硫酸镁(均为分析纯)上海国药集团化学试剂有限公司;EMR-Lipid 安捷伦科技(中国)有限公司;Cleanert LipoNo 天津博纳艾杰尔科技有限公司;实验用水为Milli-Q高纯水。
1.2 仪器与设备
Dionex Ultimate 3000超高效液相色谱仪、TSQ Quantiva 三重四极杆质谱仪(配有电喷雾离子源(electrospray ionization,ESI))美国Thermo Fisher Scientific公司;VORTEX 2涡旋混匀仪 德国IKA公司;R-300型旋转蒸发仪 瑞士Büchi公司;Avanti JXN-26高速冷冻离心机 美国贝克曼库尔特有限公司。
1.3 方法
1.3.1 标准溶液配制
扑草净、DIP、DDIP、扑草净亚砜、2-羟基-扑灭津标准储备液(100 µg/mL):分别准确称取扑草净、DIP、DDIP、扑草净亚砜、2-羟基-扑灭津标准品各5.00 mg,置于50 mL棕色容量瓶中,加入适量甲醇充分溶解后,用甲醇定容至刻度线,超声混匀备用。
扑草净-D6标准储备液(100 µg/mL):准确称取扑草净-D6标准品5.00 mg,置于50 mL棕色容量瓶中,加入适量甲醇充分溶解后,用甲醇定容至刻度线,超声混匀备用。
扑草净及其代谢混合标准中间液(1 µg/mL):分别准确移取1 mL扑草净、DIP、DDIP、扑草净亚砜、2-羟基-扑灭津标准储备液,置于100 mL棕色容量瓶中,用甲醇定容至刻度线,超声混匀备用。
扑草净-D6标准中间液(10 µg/mL):移取5 mL扑草净-D6标准储备液,置于50 mL棕色容量瓶中,用甲醇定容至刻度线,超声混匀备用。
扑草净-D6标准工作液(100 ng/mL):移取1 mL扑草净-D6标准中间液,置于100 mL棕色容量瓶中,用甲醇定容至刻度线,超声混匀备用。
1.3.2 标准曲线的制作
用20%乙腈-甲酸溶液配制系列质量浓度的混合标准溶液,每份混合标准溶液均含有扑草净、DIP、DDIP、扑草净亚砜、2-羟基-扑灭津混合外标和内标物扑草净-D6。每份混合标准溶液中内标质量浓度均为20 ng/mL,外标系列质量浓度分别为1.0、5.0、10、50、200、500 ng/mL。
1.3.3 前处理方法
1.3.3.1 样品制备
草鱼去鳞,带皮沿背脊取肌肉部分;克氏原螯虾去头、壳、肠腺,取肌肉部分;试样切成不大于0.5 cm×0.5 cm×0.5 cm小块后均质、混匀。中华绒螯蟹去壳、鳃后,将所有可食部分全部取出并绞碎混匀。所有待测样品密封标记后置于-20 ℃冷冻保存,备用。
1.3.3.2 样品前处理
称取2 g(精确至0.01 g)均质的肉糜样品,置于50 mL离心管中,加入200 μL扑草净-D6标准工作液(100 ng/mL),混匀后静置10 min,加入10 mL乙腈和3 颗玻璃珠均质子涡旋混匀30 s,随后依次加入2 g氯化钠和1 g无水硫酸镁,涡旋混合2 min后超声提取5 min,5000 r/min离心5 min,取上清液至鸡心瓶中,残渣按上述方法重复提取1 次,合并提取液,40 ℃减压蒸干。
向鸡心瓶中加入2 mL 20%甲醇溶液,涡旋混合至瓶内残留物全部溶解,将上述复溶液置于10 mL离心管中,加入0.20 g EMR-Lipid与0.05 g Cleanert LipoNo混合净化剂,涡旋振荡1 min,10000 r/min冷冻离心10 min,取1 mL上清液经0.22 μm聚砜醚滤膜过滤后,待测。
1.3.4 液相色谱条件
Thermo Hypersil GOLD C18色谱柱(100 mm×2.1 mm,1.9 µm);流动相:A为乙腈,B为0.1%甲酸溶液;流速0.3 mL/min;柱温30 ℃;进样量5 µL;梯度洗脱条件:0~1.0 min,20% A、80% B;1.0~6.0 min,20%~80% A、80%~20% B;6.0~8.0 min,80% A、20% B;8.0~8.1 min,80%~20% A、20%~80% B;8.1~10.0 min,20% A、80% B。
1.3.5 质谱条件
加热大气压ESI;正离子模式扫描;扫描模式为选择反应监测模式;喷雾电压为3500 V;蒸发温度为350 ℃;离子传输毛细管温度为330 ℃;鞘气和辅助气均为氮气,流速分别为40 L/min和10 L/min;碰撞气(氩气,纯度≥99.999%);碰撞气压力2.0 mTorr。扑草净及其代谢物的质谱检测参数如表1所示。

表1 扑草净及其代谢物质谱检测参数Table 1 Mass spectrometric parameters for prometryn and its metabolites
1.3.6 基质效应(matrix effect,ME)评估
水产品中含量较高的蛋白质、磷脂等内源性物质会因与待测物离子竞争液-液表面,从而产生基质抑制或基质增强效应,有关研究表明基质种类、基质质量和待测物质量浓度对ME均有一定影响[31],因此为评估建立方法的准确度,应考虑ME对检测结果的影响,本研究采用提取净化后添加标样法,按照下式计算ME:
基质匹配标准曲线制备:取均质的阴性草鱼、克氏原螯虾和中华绒螯蟹肌肉组织样品,不加内标物扑草净-D6,其余步骤按照1.3.3.2节进行样品前处理,制备得到不同水产品空白基质溶液。用不同水产品空白基质溶液配制系列质量浓度标准工作液,其中内标物扑草净-D6质量浓度为20 ng/mL,扑草净及其代谢物混合外标质量浓度依次为1.0、5.0、10、50、200、500 ng/mL,以此建立基质匹配标准曲线。
1.3.7 克氏原螯虾肌肉中扑草净及其代谢物残留测定
选取体质量为(26.94±6.78)g的鲜活克氏原螯虾,按9.6 µg/mL的质量浓度药浴扑草净溶液,药浴36 h后换干净爆气水继续暂养,每个实验箱每只克氏原螯虾用隔板隔开。给药后在0.5、1、2、4、6、8、12、18、24、48、72、96、120、144 h和168 h采集克氏原螯虾肌肉组织。克氏原螯虾肌肉组织均质后于-18 ℃冷冻保存,待分析。分析前将克氏原螯虾肌肉样品解冻,前处理步骤及仪器分析方法同1.3.3~1.3.5节。
1.4 数据处理
采用Trace Finder 3.2数据处理系统(美国Thermo Fisher Scientific公司)对样品中的扑草净及其代谢物进行超高效液相色谱-串联质谱分析、数据采集和处理,采用Origin 2023进行统计计算和图表绘制。
2 结果与分析
2.1 质谱条件优化
扑草净及其代谢物均为含氨基的三嗪类化合物,质谱分析时宜选择ESI正离子模式测定,在流动相中加入一定量的甲酸,不仅能够提高目标物的离子化效率,还能改善色谱峰峰形,提高色谱峰的分离度。取质量浓度为1 μg/mL的扑草净及其代谢物混合标准溶液,用蠕动泵将其随流动相一同注入质谱仪中,在ESI+模式下进行母离子全扫描,手动调试质谱参数(喷雾电压、蒸发温度、离子传输毛细管温度、鞘气和辅助气流速等)使目标物的质谱强度和稳定性达到最佳状态,然后仪器自动对目标物进行质谱条件优化。优化后的相关参数见表1。
2.2 色谱条件优化
本研究对比Thermo Hypersil GOLD C18色谱柱(100 mm×2.1 mm,1.9 µm)和Thermo Hypersil GOLD C18色谱柱(100 mm×2.1 mm,5 µm)对扑草净及其代谢物的分离效果,结果显示,粒径更小的色谱柱在峰形、分辨率、保留时间等方面表现更佳。流动相的组成直接影响分析物的分离效果和检测灵敏度[32],甲醇和乙腈是农兽药残留分析时常用的流动相[33-34],本研究考察了水相为0.1%甲酸溶液,有机相分别选用甲醇和乙腈时目标物的分离效果和质谱响应强度。如图1所示,采用甲醇时,代谢物2-羟基-扑灭津与DIP无法彻底分离,扑草净亚砜和扑草净色谱峰不仅重叠在一起,而且出峰时间位于高有机相冲洗色谱柱阶段,极易受到样品中强极性杂质干扰;采用乙腈时,虽然扑草净及其代谢物质谱强度稍有降低,但所有分析物的保留时间均提前至线性梯度洗脱阶段,并且5 个目标物的色谱峰分离度显著提升,因此选择乙腈-0.1%甲酸溶液作为流动相。

图1 扑草净及其代谢物总离子流图(50 ng/mL)Fig.1 Total ion current chromatograms of prometryn and its metabolites (50 ng/mL)
2.3 前处理方法优化
2.3.1 提取溶剂优化结果
以扑草净及其代谢物均未检出的阴性草鱼为基质,添加质量浓度为10 ng/mL的扑草净及其代谢物混标溶液,分别以甲醇、乙酸乙酯、乙腈和1%甲酸-乙腈溶液作为提取溶剂,参照1.3.3.2节进行前处理,考察不同提取溶剂对5 种目标物的萃取回收率。图2表明,当选择甲醇作为提取溶剂时,代谢物DIP和扑草净亚砜加标回收率超过120%,而代谢物DDIP和2-羟基-扑灭津回收率仅为60%~70%,由于甲醇与水互溶导致样品浓缩时不容易蒸干,从而影响实验结果的准确性;当选择乙酸乙酯提取目标物时,加标回收率可达90%~130%之间,但平行样测定结果偏差较大,结果的稳定性和重复性较差,同时乙酸乙酯容易溶解水产品中的大量脂肪,提取液颜色深且后续净化复杂;使用1%甲酸-乙腈提取时,扑草净亚砜回收率低于20%,说明扑草净亚砜在酸性环境中的萃取效率较低,从而导致加标回收率偏低;采用乙腈作为提取溶剂时,5 种目标物加标回收率达75%~105%,乙腈具有沉淀蛋白等作用,提取液颜色较浅且各目标物回收率均满足分析要求,因此选择乙腈作为提取溶剂。

图2 不同提取溶剂对扑草净及其代谢物回收率的影响(n=3)Fig.2 Effects of different extraction solvents on the recoveries of prometryn and its metabolites (n=3)
2.3.2 净化试剂优化结果
采用超高效液相色谱-串联质谱分析水产品药物残留时,与水产品共萃出的脂肪、蛋白质等杂质会影响目标物的电离能力,造成目标物信号强度的改变,因此在前处理中应尽量提高净化效果,去除杂质,避免干扰检测,污染或损害检测仪器。本研究对比净化剂Cleanert LipoNo、EMR-Lipid、Cleanert LipoNo与EMRLipid混合物对扑草净及其代谢物的净化效果,图3表明,3 组试剂净化时扑草净及其代谢物的加标回收率均可达80%~120%,Cleanert LipoNo作为一种新型除脂材料,其填料表面修饰了许多长的碳链,可针对性地吸附脂肪,使用该净化试剂时仅需混合后静置分层,无需离心,操作方便,但DIP、DDIP和扑草净亚砜回收率高于117%,基质增强效应显著。EMR-Lipid结合了体积排阻和疏水作用,可选择性去除样品中的主要脂类和大分子杂质,选用该试剂净化时,除扑草净亚砜加标回收率高于120%之外,其余目标物回收率均小于110%,其测量准确性能够满足检测需求,但平行样间的相对标准偏差(relation standard deviation,RSD)大于11%,结果的重复性较差。将两种净化试剂组合用于样品净化时,5 种目标物加标回收率为85%~106%,平行样间的RSD小于6%,说明其去除基质干扰的能力最佳。因此,选用Cleanert LipoNo与EMR-Lipid混合物作为样品前处理的净化试剂。
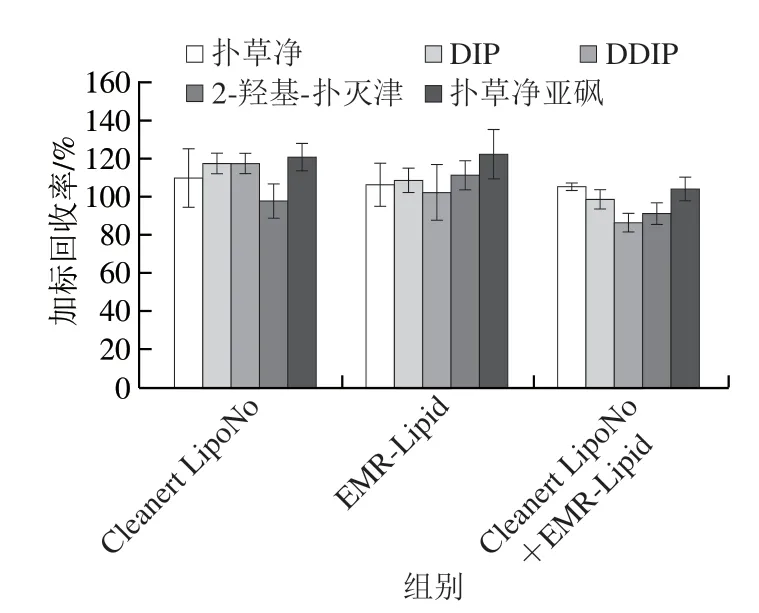
图3 不同净化试剂对扑草净及其代谢物回收率的影响(n=3)Fig.3 Effects of different cleanup sorbents on the recoveries of prometryn and its metabolites (n=3)
2.4 方法学验证
2.4.1 线性范围与灵敏度
按照1.3.2节配制标准系列工作液,以标准系列工作液中扑草净及其代谢物的质量浓度为横坐标,系列质量浓度下扑草净及其代谢物的定量子离子峰面积与内标物扑草净-D6子离子峰面积的比值为纵坐标,绘制标准曲线。结果表明,扑草净及其代谢物在1.0~500 ng/mL范围内线性关系良好,确定系数R2均大于0.992。在空白样品中加入系列低质量浓度扑草净及其代谢物混合标准溶液,以目标峰不小于3 倍信噪比(RSN≥3)确定方法的检出限(limits of detection,LOD),以不小于10 倍信噪比(RSN≥10)确定方法的定量限(limits of quantification,LOQ),得出水产品基质中扑草净及其代谢物的LOD为0.20 µg/kg,LOQ为0.50 µg/kg,方法灵敏度较高,结果见表2。
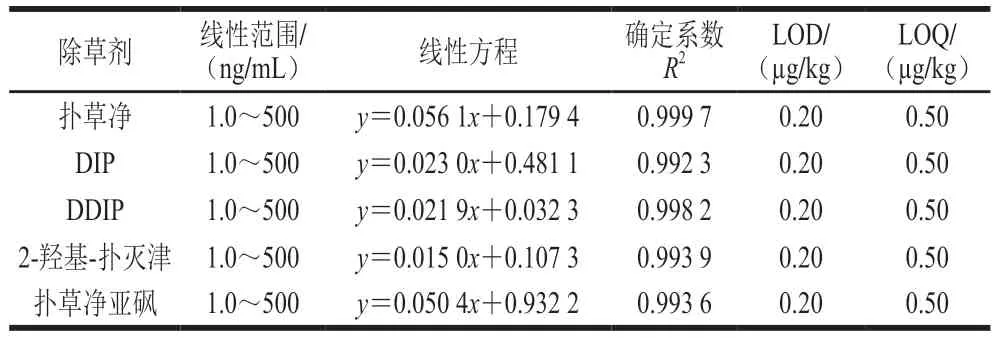
表2 扑草净及其代谢物线性范围、线性方程、确定系数、LOD和LOQTable 2 Linear ranges,linear equations,correlation coefficients,LODs and LOQs of prometryn and its metabolites
2.4.2 回收率与精密度结果
取阴性草鱼、克氏原螯虾和中华绒螯蟹肌肉组织样品,依据GB/T 27404—2008《实验室质量控制规范 食品理化检测》附录F[35]的技术要求,选取LOQ、中间浓度、常见限量值3 个水平进行加标回收实验,扑草净及其代谢物3 个添加量分别为0.5、10、50 µg/kg,每个添加量进行6 次平行实验,计算方法的加标回收率和RSD。如表3所示,草鱼、克氏原螯虾、中华绒螯蟹等不同水产品基质中扑草净及其代谢物的加标回收率为74.4%~113.7%,RSD为3.17%~11.47%,均有良好的准确度与精密度,符合农药残留分析的要求。

表3 不同水产品中扑草净及其代谢物回收率和RSD(n=6)Table 3 Recoveries and RSD of prometryn and its metabolites in different aquatic samples (n=6)
2.4.3 ME评估结果
采用基质标准曲线斜率与溶剂标准曲线斜率的比值,评价ME的影响。标准曲线法优于单点或者多点单独评价ME,更具有统计意义和代表性。当ME<0.85时,存在基质抑制效应;当ME为0.85~1.15时,ME不明显;当ME>1.15时,存在基质增强效应[36]。不同基质中扑草净及其代谢物ME结果如表4所示,结果显示,通过内标法进行校正后,5 种扑草净及其代谢物在不同水产品中的ME不明显,ME均位于0.85~1.15之间,这一结果证实了内标法可以消除ME造成的定量结果偏差,提高检测方法的稳定性和精准性。因此本研究采用溶剂标准曲线内标法定量分析5 种扑草净及其代谢物含量。
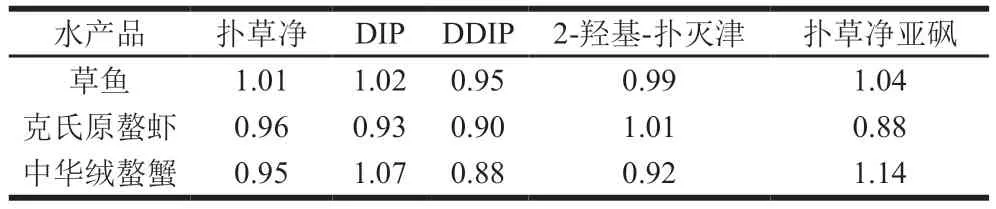
表4 不同水产品中扑草净及其代谢物的METable 4 Matrix effects of prometryn and its metabolites in different aquatic samples
2.5 克氏原螯虾肌肉中扑草净及其代谢物残留分析
应用建立的方法分析药浴扑草净后的克氏原螯虾阳性样品,以此评价方法的适用性。克氏原螯虾样品按照1.3.7节处理后,将检出目标物与标准品进行对比确认,结果显示药浴扑草净后的克氏原螯虾肌肉样品中主要检测出3 种目标物残留,其中包括扑草净及其代谢物DIP和扑草净亚砜(图4),对残留物质进行精准定性后,再利用建立方法准确评估扑草净及其代谢产物DIP和扑草净亚砜在克氏原螯虾肌肉组织中的残留水平。克氏原螯虾肌肉组织中扑草净及其代谢产物药-时曲线详见图5。可以看出,与菲律宾蛤仔对扑草净的富集规律类似[37],药浴扑草净后,克氏原螯虾肌肉中的扑草净残留量随时间大幅波动,先后出现2 个较为明显峰,不同的是菲律宾蛤仔体内扑草净残留量在24 h达到峰值,而克氏原螯虾肌肉中扑草净及其代谢物残留量在12 h达到峰值,说明克氏原螯虾对扑草净具有快速富集效应,但对比图5中扑草净原药和代谢物残留量变化趋势可以看出,在短时间内扑草净的富集效率远高于代谢效率。药浴扑草净36 h后将克氏原螯虾转移至洁净水中,其肌肉中扑草净残留量迅速下降,该结果与菲律宾蛤仔[37]和海参[38]体内的扑草净消除实验数据基本一致,说明扑草净在水中消除较快。

图4 克氏原螯虾肌肉组织中扑草净及其代谢物选择反应监测色谱图Fig.4 SRM chromatograms of prometryn and its metabolites in muscle tissue of crayfish
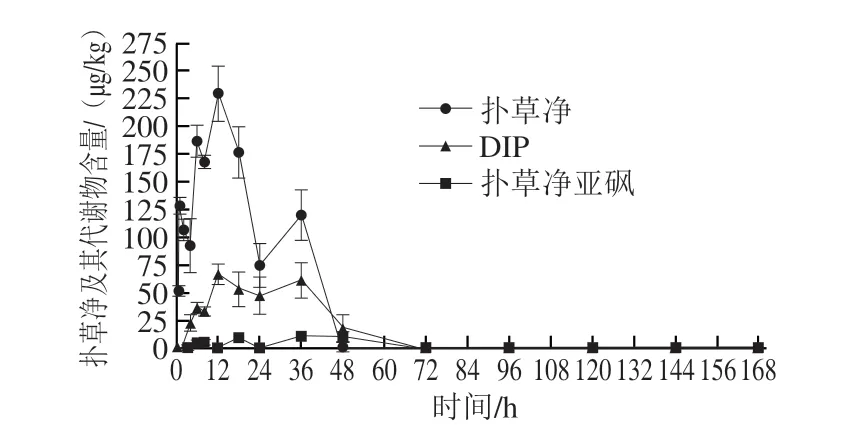
图5 克氏原螯虾肌肉组织中扑草净及其代谢物药-时曲线(n=5)Fig.5 Drug-time curves of prometryn and its metabolites in muscle tissue of crayfish (n=5)
3 结论
本研究开发了一种水产品中扑草净及其代谢物残留的QuEChERS-超高效液相色谱-串联质谱检测方法,样品经乙腈提取,EMR-Lipid与Cleanert LipoNo组合净化,甲醇溶液定容后超高效液相色谱-串联质谱测定,内标法定量。扑草净及其代谢物在1.0~500 ng/mL范围内线性良好,确定系数R2大于0.992。5 种除草剂LOD为0.20 μg/kg,LOQ为0.50 μg/kg。扑草净及其代谢物在3 种水产品基质中的加标回收率为74.4%~113.7%,RSD为3.17%~11.47%。该方法操作简单快速、基质干扰少、灵敏度高,可实现水产品中扑草净及其代谢物残留的精准识别和准确定量,为扑草净在水产动物中的药代动力学和残留消除规律研究提供理论依据和技术支撑。
