一个新发SERPINF1基因突变的Ⅵ型成骨发育不全病例家系分析
2024-03-01谢泽慧刘琳毛斌郭亚荣田琦民马晓玲
谢泽慧,刘琳,,毛斌,郭亚荣,田琦民,马晓玲,*
(1.兰州大学第一临床医学院,兰州 730000;2.兰州大学第一医院生殖医学中心,兰州 730000)
成骨发育不全(osteogenesis imperfecta,OI)又称脆骨病-蓝巩膜-耳聋综合征,是由于编码Ⅰ型胶原蛋白的基因发生突变从而导致的广泛性结缔组织疾病[1-3],是一种以骨骼发育不全和骨脆性增加为主要特点的单基因遗传病[4],发病率约为1/15 000~1/20 000[5],是严重影响新生儿健康的出生缺陷疾病之一。有85%~90%的OI病例为常染色体显性遗传[5]。近年来随着分子遗传学的发展,研究者们发现了19种导致OI的致病基因,其中常染色体隐性遗传基因12种(SERPINF1、CRTAP、P3H1、PPIB、SERPlNH1、FKBP10、SP7、BMP1、TMEM38B、WNT1、CREB3L1、PLOD2、P4HB、SEC24D、SPARC等)[6]。SERPINFI突变型OI的特征表现为轻微外力下的反复骨折、长骨屈曲畸形、骨质发育不良等[6],随着疾病进一步发生发展,可能导致患者脊柱后、侧凸及四肢严重畸形[7],除此之外,还有蓝色巩膜、牙质发育不全和听力损失等骨外表现[8]。
1979年,Sillence等[9]根据患者的临床表现、影像学特征和遗传模式将OI分为Ⅰ~Ⅳ型,之后的研究者们又根据OI的致病机制及基因的不同将OI的分型扩展为16型[10]。其中,SERPINF1基因编码的色素上皮衍生因子(pigment epithelium-derived factor,PEDF)调节成骨细胞及破骨细胞活性、影响骨骼矿化,PEDF水平低下会导致骨矿化不足和矿化时间延长[11],这一类OI在分类上被归为Ⅵ型。Ⅵ型OI往往于儿童期发病,存在中至重度骨骼畸形[12],可见类骨、“鱼鳞样”板层骨的外观[13],伴或不伴有蓝色巩膜,光镜下可见骨质鱼鳞样结构伴矿化延迟,严重程度介乎于I型(轻型)和Ⅲ型(存活患者中最严重类型)之间[14-17]。目前广为认可的治疗OI患者最有效的药物是双磷酸盐(bisphosphonates,BPs)[18],可以增加骨密度、降低骨折率、有效改善患者生存质量,但是该类药物无法改善骨矿化不足,对Ⅵ型OI的治疗效果不佳[19]。近年来研究显示,RANKL抗体狄诺塞麦、骨硬化素单抗(Scl-Ab)和转化生长因子β中和抗体等新型药物对于Ⅵ型OI有着良好的治疗前景[20]。
本文针对1例SERPINF1基因同义突变导致的Ⅵ型OI家系进行全外显子组基因测序,明确该家系的致病基因,分析其基因型、致病模式和基因型-表型相关性。
资料与方法
一、研究对象
1.家系来源:该OI家系由兰州大学第一医院生殖医学中心采集,包括先证者、同胞弟弟和父亲,母亲因去世未能采集。血样以及临床资料的收集均符合知情同意原则。本项研究已通过兰州大学第一医院生殖医学伦理委员会的审查批准(批准文号为LDYYSZLL2023-17)。
2.试剂及仪器:核酸纯化试剂盒(Agencourt Ampure XP-Medium kit)、RNA提取试剂盒及反转录试剂盒均购自深圳华大基因;Agilent 2100 Bioanalyzer分析仪(Agilent DNA 1000 Reagents,安捷伦,美国)、荧光定量仪(Qubit Fluorometer,BMG Labtech,德国)、MGISEQ-2000测序平台(PE100/PE150,武汉华大智造科技有限公司)。
二、DNA提取和全外显子测序
在进行助孕前遗传咨询时,建议患者及其父母、弟妹接受基因检测,经过医院伦理委员会审查并取得患者同意后,对患者、患者父亲和患者弟弟各采外周血2 ml,样本送交深圳华大基因检测中心测序。从受检者血液中提取基因组DNA,并打断制备文库,然后通过Roche KAPA HyperExome芯片对目标基因外显子及临近剪切区的DNA进行捕获和富集,最后使用MGISEQ-2000测序平台进行变异检测(测序数据质控指标为:目标区域平均测序深度≥180×,其中目标区平均深度>20×的位点所占比例>95%)。测序片段通过BWA与UCSC hg19人类参考基因组进行比对,去除重复。使用GAT进行碱基质量值校正SNV、INDEL和基因型检测。使用ExomeDepth进行外显子水平的拷贝数变异检测。测序由深圳华大基因检测中心完成。
三、Sanger测序验证
使用公共数据库信息,查询NCBI、UCSC或Ensembl数据库上待测基因的序列信息,确定待测基因在染色体上的定位和编码区大小,然后使用Primer软件对其目标区域的外显子编码区及其侧翼序列进行引物设计,在引物设计完成以后使用在线的电子PCR模拟进行特异性检测和验证,PCR扩增体系使用2×TaqMastermix扩增。扩增完成后使用琼脂糖凝胶电泳,测序完成后使用DNAstar软件中的SeqMan程序将测序结果与参照序列进行比对,分析测序结果。引物序列及扩增产物信息详见表1。
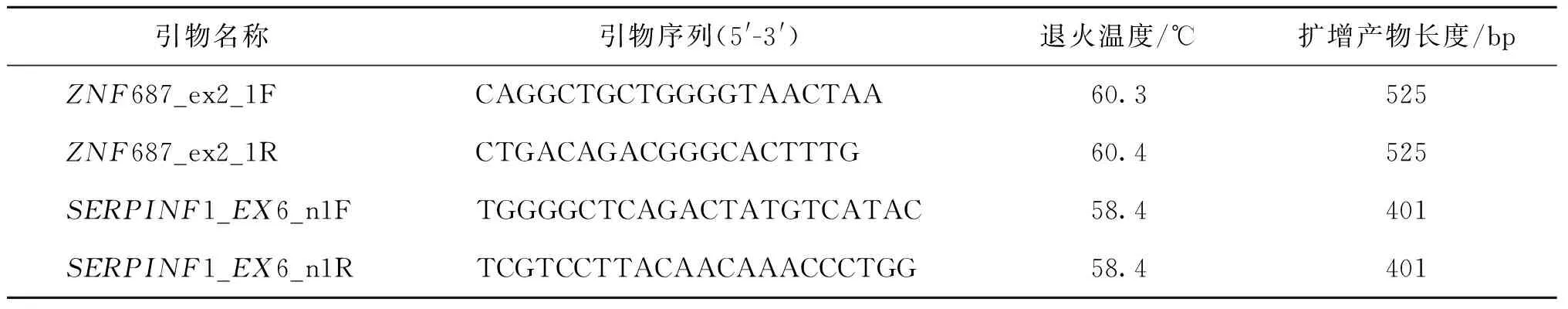
表1 引物序列、退火温度及扩增产物信息
结 果
一、病例资料分析
患者,女,34岁,疑似患有先天性成骨不全,2021年求助于兰州大学第一医院生殖医学中心希望通过辅助生殖技术生育1名健康后代。该患者身高1.2米,体重50 kg,结婚5年,婚前与前任因个人因素流产1次,婚后工具避孕2年,后未避孕未孕3年。自述3岁前无明显表型,3~15岁期间易发骨折,尤其近肢易发,多次行手术治疗,15岁后易发骨折症状消失至今,可蹲距行走,但近5年行动受限,出行需依靠轮椅。患者站立困难,查体见三角脸,脊柱侧弯畸形,胸廓前凸,无智力障碍。该患者虽曾存在多发易发骨折情况,且体格检查可见站立困难、脊柱侧弯畸形等与OI高度符合的临床症状,但患者未进行过完整的临床评估及基因检测来确诊OI。患者母亲早年因疾病去世(非相关疾病),父亲体健,其弟弟具有相似临床表现,妹妹体健且婚后育有1健康男婴,否认家族近亲结婚史、家族遗传病史及致畸因素接触史。患者于2021年8月在我院行子宫输卵管造影示:左侧输卵管通而欠畅,右侧输卵管通而不畅,盆腔轻度粘连。其丈夫2021年6月于我院行精液常规检查示:精子浓度8.97×106/ml,前向运动精子21%,形态正常精子百分数1.35%,精子DNA碎片指数5.72%。
生殖中心经过综合评估后建议患者夫妇放弃妊娠,但患者及其家属因生育需求强烈要求辅助生殖助孕,于是充分告知患者辅助生殖技术失败风险及成功妊娠后对母儿可能造成的不良后果等。患者及其家属表示充分知情同意,愿意承担妊娠及分娩的一切相关风险及责任,并签署知情同意书。
二、影像学结果分析
先证者全脊柱正侧位片及胸部X光片显示其有明显胸廓畸形,双侧胸廓不对称,呈现左低右高改变,气管、纵膈左移,上纵膈增宽,肋骨走形扭曲,脊柱生理弧度消失,胸段向后突出,明显向左侧弯,椎体骨质边缘欠光整。骨盆不对称,双侧股骨干侧弯,双侧骶髂关节未见明显异常,双侧股骨上段及股骨头形态、结构正常(图1)。
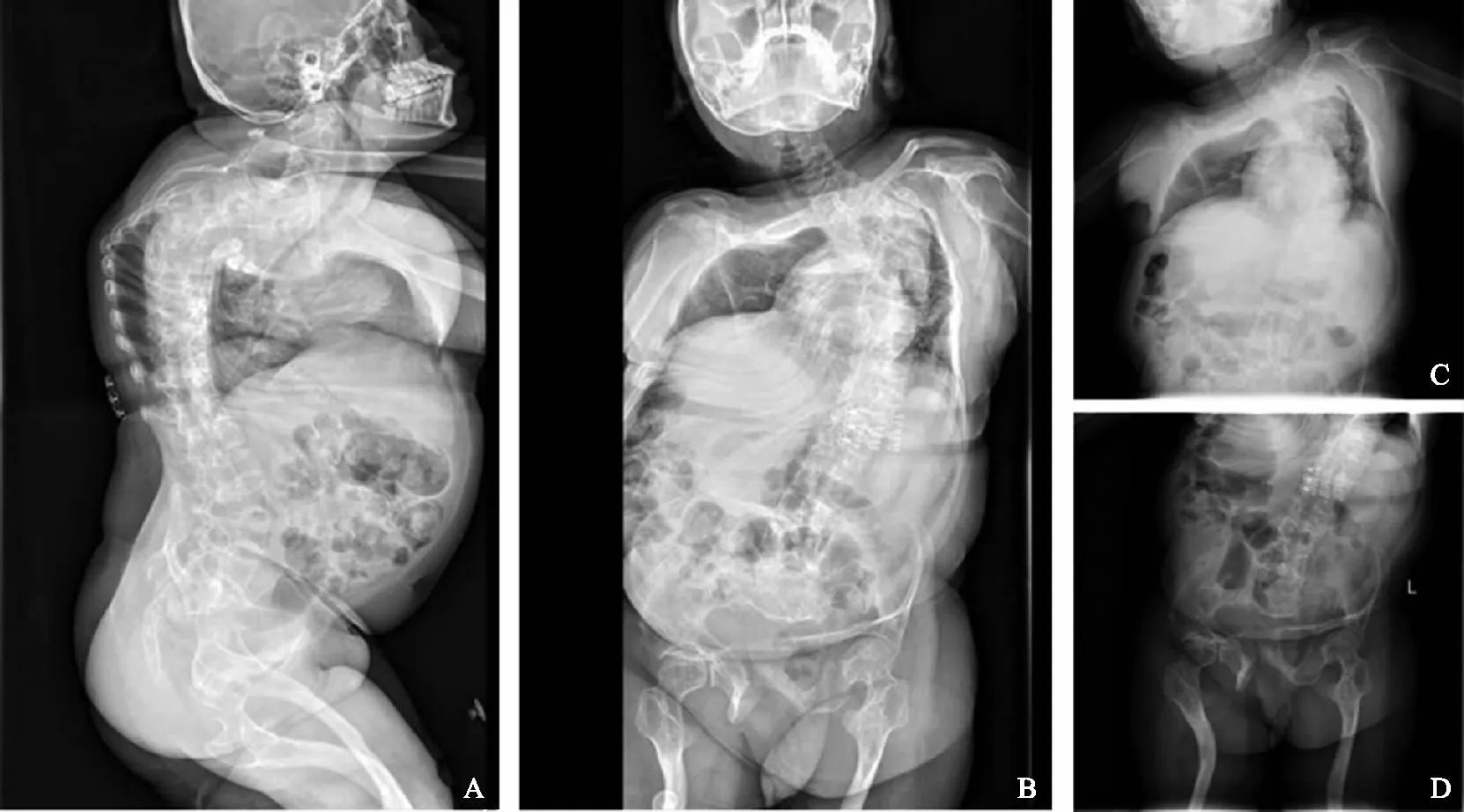
A:侧面,脊柱胸段向后突出;B:正面,胸段明显向左侧弯;C:胸廓呈左低右高改变;D:骨盆不对称,双侧股骨干侧弯。图1 先证者的影像学检查结果
三、全外显子组测序结果
基因检测结果显示:先证者及其弟弟在SERPINF1基因上chr17:1678494(NM_002615.5)位点发生了突变c.786G>A,此突变并未导致氨基酸发生改变p.Lys262Lys,故该突变为同义突变。目前没有该变异致病性的相关报道,根据美国医学遗传学与基因组学学会(ACMG)指南(附录),该变异被判断为意义未明变异。先证者及其弟弟基因型为纯合突变,其父亲为该致病基因的杂合携带者,根据该基因常染色体隐性遗传的遗传模式推测其过世的母亲亦为该基因的杂合携带者,患者丈夫基因检测结果正常。家系图如图2所示。

图2 家系图(Ⅱ-2为先证者)
利用ESP数据库(http://olap.epsnet.com.cn/guidedetail1.html)、千人数据库(https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/data/)、EXAC数据库(http://exac.broadinstitute.org)检索发现该突变为正常对照人群中未发现的变异(或隐性遗传病中极低频位点),通过保守性预测、进化预测、剪接位点影响等多种统计方法预测出该变异会对基因或基因产物造成有害的影响。
四、Sanger测序结果
为检测c.786G>A位点对RNA剪切的影响,并明确先证者的助孕策略,提取先证者夫妻的RNA,经反转录得到cDNA后,利用引物进行PCR扩增,Sanger测序后将测序结果使用DNAstar软件中的SeqMan程序比对参照序列。通过和参照序列比对可以看出先证者(Ⅱ-2)和其弟弟(Ⅱ-3)在SERPINF1基因上的NM_002615.5序列发生了c.786G>A的纯合突变,其父亲(I-1)在此处为杂合突变,但是均未导致该处赖氨酸的改变,属于同义突变。而表型正常的先证者丈夫在此处并未发生改变(图3)。
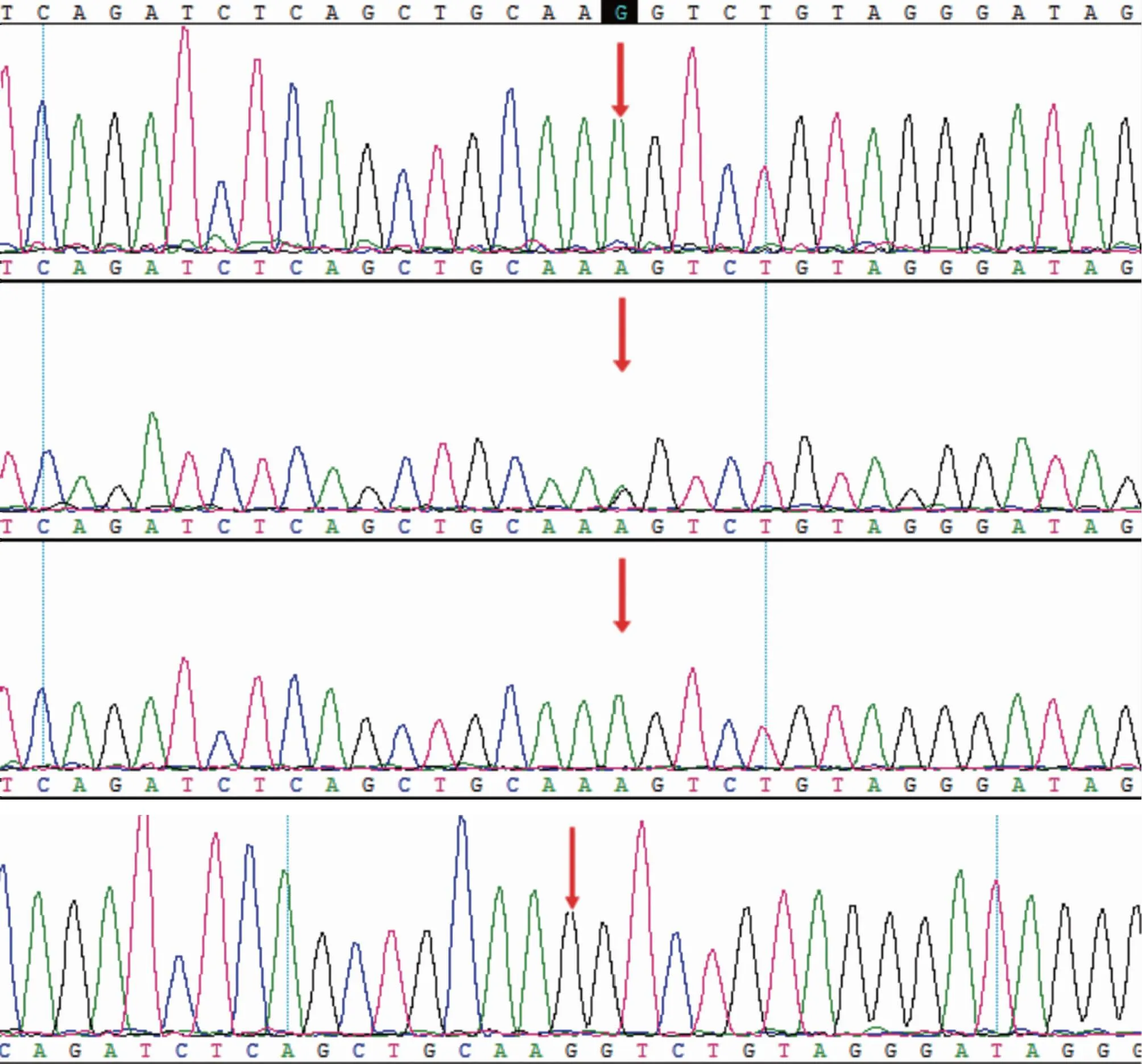
图3 SERPINF1位点的Sanger测序峰图[由上至下分别为先证者(Ⅱ-2)、其父(Ⅰ-1)、其弟(Ⅱ-3)、其丈夫(Ⅱ-1)]
五、辅助生殖结果
患者为该致病基因的纯合携带者,其丈夫不携带该致病基因,因此夫妻双方的子女必定为该致病基因携带者但不发病,因此在综合评估该患者情况后认为无需进行胚胎植入前遗传学诊断(preimplantation genetic testing,PGT),最后确定使用体外受精-胚胎移植(IVF-ET)方法为其助孕。结合患者夫妻各项情况后使用拮抗剂方案促排卵,取卵11枚,获得可移植胚胎3枚,全胚冷冻保存。2022年1月激素替代周期准备内膜后移植冻融胚胎1枚未孕,1个月后再次移植冻融胚胎1枚,患者成功怀孕,之后随访患者宫内单胎妊娠,孕34+2周于本院剖宫产1女,女儿体健。
讨 论
自2002年Ⅵ型OI首次被报道[21]以来,该类型疾病受到了更多的关注。近年来随着基因测序技术的不断发展,遗传基因突变的检测和对其编码蛋白质功能和结构的预测等技术逐渐成熟,对于Ⅵ型OI的基因诊断也步入了新阶段。本文报道了1例由于SERPINFI基因chr17:1678494(NM_002615.5)发生同义突变导致OI的家系,该突变位点此前从未被报道过,通过对该位点致病的研究,分析其对机体相关组织结构和功能的影响,预测其致病性,丰富了Ⅵ型OI的表型。
OI是一种由于基因突变导致Ⅰ型胶原蛋白发生含量或结构改变[22]所引起的一种单基因遗传疾病,主要特点为骨量减少和骨骼脆性增加,常表现为反复轻微外力下骨折和骨骼畸形[23]。OI多数表现为常染色体显性遗传[6],近年来常染色体隐性遗传和X染色体遗传等遗传模式也被报道[22]。本文中导致先证者及家系致病的基因SERPINF1即为常染色体隐性遗传模式,该基因位于染色体17p13.3,它与OI常见致病基因COL1AI和COL1A2影响成骨细胞的胶原形成和分化不同,而是会增加骨吸收。SERPINF1基因编码PEDF蛋白,该蛋白通过RANK-RANKL-OPG途径上调骨保护素(osteoprotegerin,OPG)的表达,通过阻断RANKL来抑制破骨细胞成熟;当SERPINF1基因突变时,由于RANK-RANKL-OPG途径的错误调节,破骨细胞分化与激活增加,导致PEDF含量下降,OPG浓度降低,破骨细胞数目增加,骨骼矿化不足,骨降解增加,从而导致了Ⅵ型OI的发生[24]。Ⅵ型OI患者的骨骼脆性及畸形程度从生活质量不受影响到严重身体机能不全不等[25],患者通常没有围产期骨折,第1次骨折一般发生在出生后4~18个月,之后的骨折频率和严重程度是循序渐进的,多不伴有蓝色巩膜、听力障碍和牙质发育不全等骨骼外表现。这使得RANKL抗体狄诺塞麦获批用于成人的骨质疏松症,并且对于SERPINF1基因突变引起的儿童Ⅵ型OI也有较好作用。
在该家系中,先证者和其弟弟SERPINF1基因纯合突变,幼时曾反复骨折,成年后有站立困难、脊柱侧弯畸形和胸廓前凸等典型临床症状,其父亲为该突变的杂合子携带者;根据该基因的遗传模式推测先证者已过世的母亲同为该突变杂合子携带者,通过该家系的遗传模式和临床发病情况分析,我们认为这种基因突变是致病的,可能导致中重度的Ⅵ型OI。
本研究在该家系中发现了两名由于SERPINF1基因突变导致的Ⅵ型OI患者及1名杂合携带者,此突变为一个从未被报道的新突变,在任何公开数据库中未能检索到该突变的报道。对该家系进行了基因检测和分析,由于该先证者求助于我们生育健康后代,根据基因测序结果为她提供遗传咨询和助孕建议,最终先证者生育了1名推测携带该变异基因但是并不发病的健康女婴;要让该突变基因在此家系中完全阻断,这名女婴在未来的生育中仍然需要进一步遗传咨询和干预。
本文对于此家系的研究仍然存在一定局限性,首先,我们并未取得先证者母亲的血样,没有对包括先证者祖父母在内的所有家族成员进行基因测试,无法绘制出完整的遗传模式图;另外,我们暂时没有对该新发突变进行功能验证和进一步实验研究;此外,Ⅵ型OI在全球的报道病例十分稀少[26],因此无法严格确定其基因型-表型的相关性,该基因突变对于临床表现的影响无法严格界定。本病例的结论并不具有完全的代表性,但是该病例的报道也进一步补充了该类疾病的变异位点和临床表型等资料。该基因突变的致病性分类为“意义未明变异”。因此在未来的研究中,不论是功能试验、家系研究还是动物模型的建立,都需要更深一步探索该基因突变的意义和致病性。
综上,本文报道了1例纯合SERPINF1基因c.786G>A(p.Lys262Lys)突变的患者通过辅助生殖技术成功妊娠的临床资料以及她的家系资料,该突变为同义突变,同义突变既往常常被认为是不致病的,但近年来的研究显示其可能具有致病性,在未来的单基因遗传病研究中需要被重视。本文丰富了OI的表型,补充了人类SERPINF1基因的突变数据库,为进一步研究Ⅵ型OI的基因型-表型相关性和未来对于此疾病的遗传咨询等提供了新的依据。
