PARK2介导的帕金森病中帕金蛋白对α-突触核蛋白的作用
2024-02-22肖琪樊慧杰李艳荣范丽珊和璐璐谢良骐贾璐王青肖保国马存根柴智
肖琪 樊慧杰 李艳荣 范丽珊 和璐璐 谢良骐 贾璐 王青 肖保国 马存根 柴智
(1山西中医药大学多发性硬化益气活血重点研究室 神经生物学研究中心,山西 晋中 030619;2山西医科大学基础医学院;3复旦大学华山医院神经病学研究所)
帕金森病(PD)是常见的神经退行性疾病之一,主要表现为运动障碍,伴有主要运动症状,如运动迟缓、僵直性和静息性震颤,还可能表现为多种非运动症状,如自主神经功能障碍、认知障碍、睡眠障碍和神经精神症状〔1〕。其特征是黑质致密部(SNpc)中含有神经黑色素的多巴胺能神经元的进行性缺失〔2〕。研究表明,在工业化国家,60岁以上人群的发病率为1%,80岁以上人群的发病率为3%以上〔3,4〕。5%~10%的PD患者有孟德尔遗传的单基因变异,其余的90%~95%是原因不明散发性变异〔5〕。Parkin基因(PARK2)突变是常染色体隐性PD最常见的病因,占早发性PD(EOPD)的50%〔6〕。Parkin是一种多效性神经保护蛋白,参与调控细胞周期、线粒体动态平衡和能量代谢等细胞进程〔7〕。PD组织学特征为路易小体的神经元内嗜酸性蛋白内含物,α-突触核蛋白(syn)是路易小体的主要成分〔8〕。本文围绕PARK2介导的PD中Parkin与α-syn在翻译后修饰、tau病理和微聚物的形成、细胞凋亡等4个方面的作用及一些PARK2患者死后的神经病理学检查进行综述。
1 Parkin
PARK2的基因编码蛋白质产物为Parkin,由12个外显子和465个氨基酸组成〔9〕。Parkin的功能丧失突变是家族性PD的主要遗传原因。PARK2基因突变导致PD的常染色体隐性形式,是EOPD最常见原因。在EOPD相关基因的筛查中对基因位点6q25的鉴定发现,2~27位点负责EOPD,之后的研究中,确定了其编码为Parkin的新基因PARK2〔10〕。Parkin作为E3-泛素连接酶发挥作用,维持泛素蛋白酶体系统清除错误折叠和聚集的蛋白质,调节线粒体自噬以防止线粒体功能障碍,并以直接和间接功能防止氧化应激〔11~15〕。
在PARK2基因的12个外显子中已经鉴定出100多个PD相关突变,包括错义突变、大染色体缺失和重复、截断突变和启动子突变。PARK2基因的纯合和复合杂合突变是遗传EOPD的原因。EOPD的临床特征,如发病年龄早、伴有昼夜波动的PD、对左旋多巴的良好反应、肌张力障碍、反射亢进、无痴呆和相对良性的病程〔16〕。杂合子错义突变可能易患晚发性PD,类似于散发性PD,但其如何及是否对PD的病理生理学起作用仍不清楚〔17〕。
除了损害Parkin蛋白功能的突变外,翻译后修饰(PTM),如S-亚硝基化、多巴胺共价结合、应激激活激酶c-Abl的磷酸化和氧化应激已被证明会损害散发性PD中Parkin的活性〔18~20〕。PD相关PARK2突变的携带者与散发性PD患者的临床症状非常相似。PARK2突变相关的独特临床表现为发病年龄较小、频繁的肌张力障碍和反射亢进,发病较早但疾病进展较慢且病程表现为良性〔21〕。治疗上,PARK2介导的PD患者对左旋多巴治疗反应良好,但容易出现左旋多巴导致的运动障碍〔22,23〕。病理上,PARK2介导的PD患者和小鼠模型中显示SNpc的神经元明显减少,蓝斑神经元中度下降〔24,25〕。
2 α-syn
α-syn是一种小的、可溶的140个氨基酸蛋白,位于突触前末端,在大脑中高度表达,特别是在一些PD影响的区域,如海马、嗅球、SNpc、迷走神经背侧运动核、外侧和内侧乳头核〔26〕。α-syn可以在各种因素下错误折叠和聚集,包括钙浓度升高,氧化应激,基因突变及与许多其他蛋白质的相互作用。随着PD进展,路易小体病理逐渐影响神经系统的更多方面。研究发现,错误折叠和聚集的α-syn可以传播到相互连接的神经元,导致内源性α-syn的聚集和错误折叠,如朊病毒蛋白〔27〕。
PD发病机制中的α-syn毒性相关的多种功能失调途径有功能失调的突触小泡运输、线粒体功能受损、内质网和高尔基体功能缺陷、自噬溶酶体途径缺陷和核功能障碍〔28〕。与野生型α-syn相比,α-syn和PTM,如丝氨酸(pSer)129α-syn的PD相关突变及氧化应激增加了其核定位〔29〕。在核内,α-syn在PD患者体内、体外和脑组织中均能结合线粒体生物发生的标志物过氧化物酶体增殖物激活受体γ激活因子(PGC)1α启动子,导致PGC1α启动子活性降低,PGC1α mRNA和蛋白质水平降低〔30〕。因此,α-syn的核定位可能导致线粒体功能障碍,是PD的主要标志之一,并损害其他可能导致PD发病的途径。研究发现,外源性α-syn导致Parkin依赖性线粒体自噬机制紊乱,使有缺陷的线粒体积累,并且Parkin的过度表达改善了α-syn寡聚物引起的线粒体功能障碍〔31〕。
3 Parkin对α-syn功能的作用
3.1Parkin影响α-syn的翻译后修饰 如图1所示,α-syn经历广泛的PTM,包括磷酸化、泛素化、截断和硝化,其中许多已经在路易小体中鉴定出来,这表明这些修饰可能是必要的,并且在α-syn聚集和神经毒性中起重要作用〔32〕。有研究〔33〕指出,Parkin功能的丧失延缓了α-synA30P突变(hA30Pα-syn)转基因小鼠的感觉运动障碍,略微改善了PD终末期神经病理学表现。hA30Pα-syn小鼠的主要神经病理学特征为含有原纤维α-syn的神经元沉积物在S129处磷酸化(PS129α-syn),PS129在D135处截断α-syn,表明了Parkin功能丧失使PS129α-syn经历了PTM。研究发现,α-syn的pSer129磷酸化与PD多巴胺能神经元细胞死亡有关〔34〕。PP2A和polo样激酶(PLK)2是pSer129α-syn的主要调节因子。含有B55的蛋白磷酶(PP2)A亚型已被证明是使pSer129α-syn去磷酸化的主要酶〔35〕。研究表明,Parkin的表达减弱了细胞死亡和炎症,降低了PLK2和糖原合成激酶(GSK)3β水平,增加了PP2A表达,导致pSer129α-syn和磷酸化tau水平降低,防止蛋白质聚集和神经原纤维缠结,表明了Parkin对抗α-syn毒性和tau过度磷酸化的重要生理功能〔33〕。研究发现,单胺氧化酶(MAO)-B的过表达导致α-synTyr39的3-硝基酪氨酸增加氧化应激(OS),导致其寡聚化。Parkin抑制MAO-B的转录和表达,Parkin缺失时,MAO-B的转录增加并相应地增加OS,而野生型Parkin慢病毒介导的表达能够降低MAO-B表达和活性〔36〕,表明Parkin通过降低MAO-B活性以防止α-syn发生硝化、寡聚化和氧化应激,并减少Tyr39硝化α-syn和低聚物的形成。
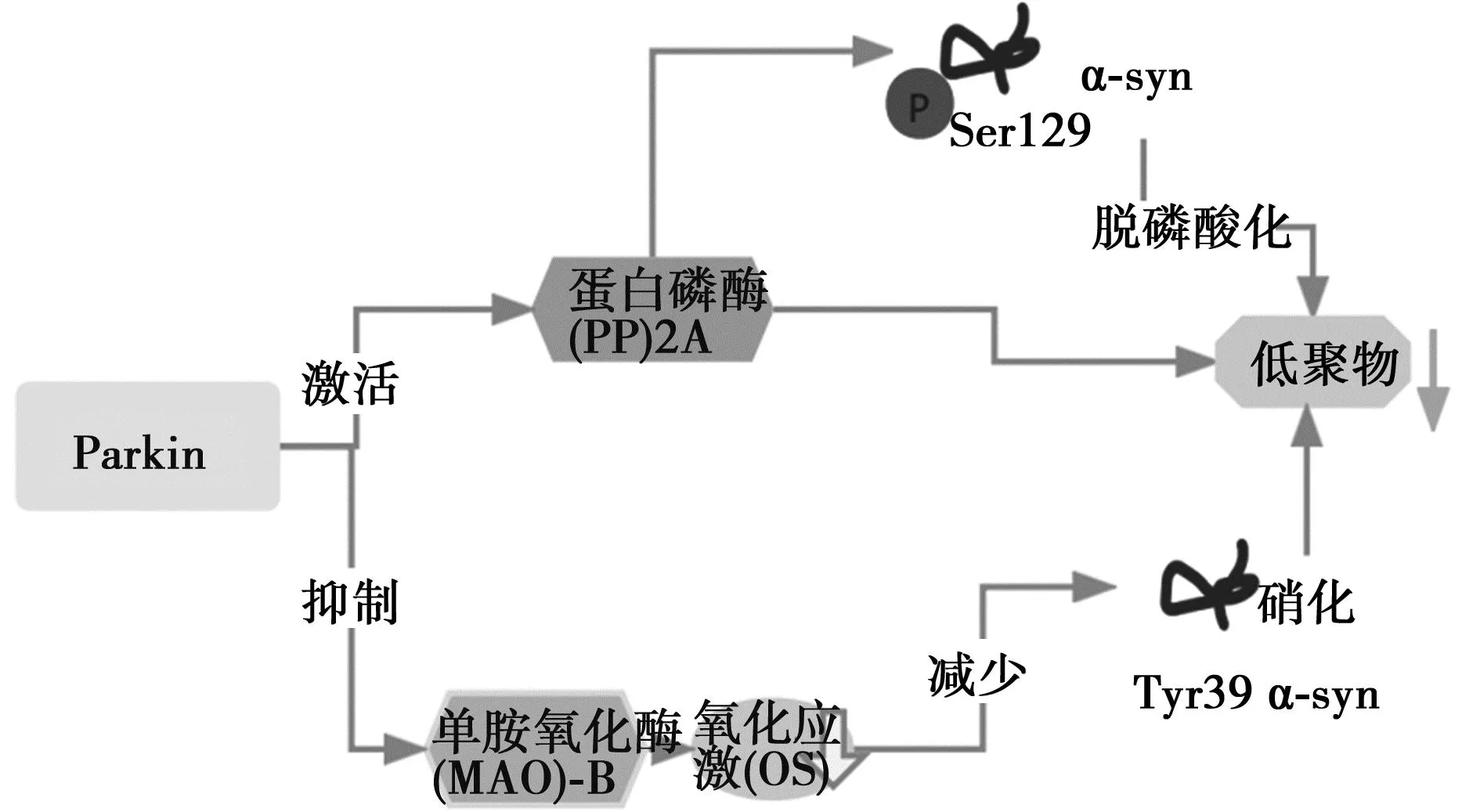
图1 Parkin影响α-syn的翻译和修饰
3.2Parkin在α-syn介导的tau病理中作用 路易小体中聚集的α-syn是PD的神经病理学标志,而神经原纤维缠结中聚集的微管相关tau是阿尔茨海默病和额颞叶痴呆的共同特征〔37〕。研究表明α-syn刺激位于微管结合区的tau蛋白激酶(PK)A介导的Ser262磷酸化,导致微管失稳和神经毒性。此外,使用1-甲基-4-苯基-1,2,3,6-四氢吡啶(MPTP)的细胞模型显示,α-syn的水平对磷酸化Ser262的数量至关重要〔36〕。特别是GSK3β已被证明是以α-syn依赖性方式在残基Ser262,Ser396和Ser404处tau中过度磷酸化的关键蛋白。GSK3β酪氨酸(Tyr)216依赖性磷酸化是其激活所必需的〔38〕。PARK2介导的PD中Parkin蛋白通过减少激活相关Tyr216处GSK3β的磷酸化,从而抑制Ser262,Ser396和Ser404处tau过度磷酸化。Parkin对PP2A的激活也会引起tau的去磷酸化,见图2。α-syn、tau、GSK3β、PP2A、PLK2和Parkin之间存在联系,是PARK2介导的PD的潜在疾病机制,证明了Parkin在α-syn和tau病理学在神经保护中的重要作用〔36〕。Parkin及其神经保护功能在PARK2介导的PD中的丧失可能导致蛋白质错误折叠,积累和聚集。
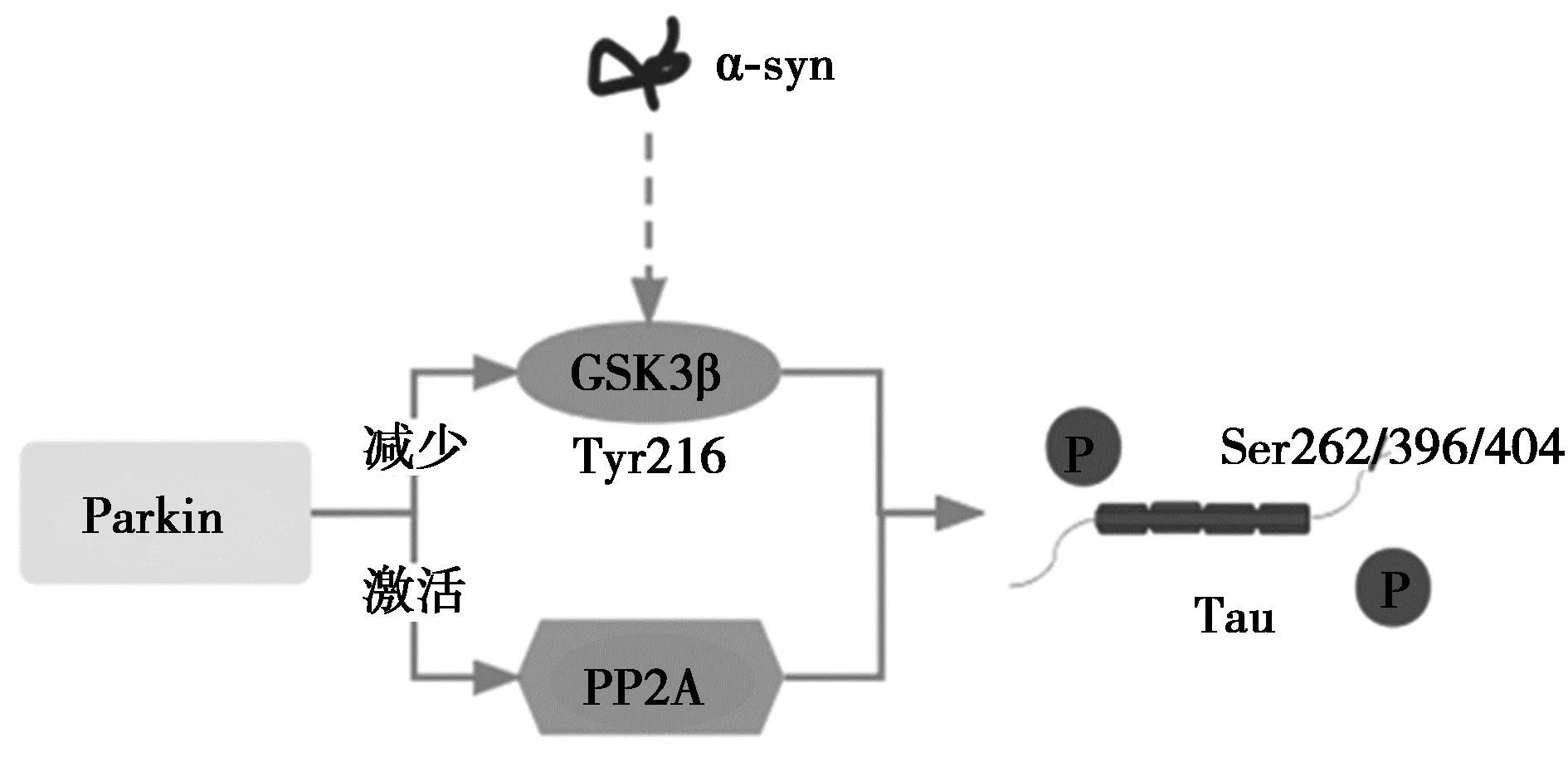
图2 Parkin影响α-syn介导的tau病理
3.3Parkin功能障碍导致α-syn微聚体形成 Parkin作为E3-泛素连接酶的关键作用是通过蛋白酶体或自噬系统介导蛋白质降解,表明Parkin在清除聚集和错误折叠的蛋白质方面具有重要功能。蛋白酶体系统对细胞内蛋白质〔如α-syn、tau和β-淀粉样蛋白(Aβ)〕积累和聚集的功能失调可能与神经退行性疾病的发生有关〔39〕。研究表明,防止α-syn、tau和Aβ过度表达及蛋白酶体抑制的保护作用可以由Parkin介导〔40〕。Parkin过表达可导致蛋白酶体酶活性增加,减少α-syn路易小体的表达,减轻α-syn导致的毒性〔41〕。这些研究表明了Parkin在增加蛋白酶体系统并降解错误折叠和聚集的蛋白质方面的重要功能。由于路易小体中存在泛素化的α-syn,提示α-syn和Parkin之间的直接相互作用可能是由蛋白酶体系统引起的α-syn降解〔42〕。然而,只有O-糖基化的α-syn被证明是Parkin底物〔43〕。Parkin间接影响α-syn的聚集和积累,可能会减弱α-syn介导的毒性。研究发现,α-syn过度表达相关的毒性,可以通过蛋白酶体抑制来模拟,可以通过Parkin的过度表达来减轻〔44〕。如表1所示,在17篇〔45~61〕PARK2患者尸检脑切片报告病例中发现,其中15篇〔45,46,48~50,52~61〕为纯合子或复合杂合子,其余2篇〔47,51〕为杂合子。其中8例报告了路易小体存在〔45~52〕,除1例为嗜碱性路易小体样包涵体外,其余7例均为典型路易小体。10例路易小体阴性的病例中发现患者平均发病年龄为25.5岁,低于7例PARK2相关病例存在α-syn和路易小体病理学患者的46.3岁〔53~60〕。2015年,1例秘鲁家族EOPD尸检病例显示SNpc中有严重的神经元丢失,纹状体中有酪氨酸羟化酶阳性纤维丢失,但未观察到路易小体病理学和α-syn阳性内含物,发现了神经原纤维缠结〔61〕。在与PARK2介导的PD中与路易小体相关的文献中存在以下几种假设:①在验尸研究中观察到的路易小体代表了偶然的路易体病理学,因为它们经常在健康的老年人中发现;②发病晚期患者的蛋白质清除系统发生功能障碍〔62〕;③一些Parkin突变可能导致残留的Parkin活性,导致路易小体形成的可能性增加。因此,关于发病年龄较小的PARK2患者可能具有更有效的蛋白质清除系统或处理异常积累蛋白质的不同机制。因此,死后神经病理学检查发现神经元丢失和胶质增多症,但没有路易小体这一现象可能是正常的。
细胞培养研究表明,Parkin通过路易小体中蛋白质的K63连锁泛素化参与形成凝集体样包涵体〔63〕。特定的错义突变,例如导致残留Parkin活性的R275W,会增加路易小体形成的可能性,而导致Parkin功能完全丧失的外显子缺失使路易小体难以形成。PTM可加速pSer129从α-syn单体向有毒低聚物和原纤维的转化〔64〕。研究发现,Parkin介导的包涵体形成是一种神经保护作用,可避免α-syn释放到细胞外空间,从而防止pSer129α-syn原纤维扩散到相互连接的神经元〔65〕。Espay等〔66〕提出了作为PD潜在机制的蛋白质聚集的三个概念框架。首先,α-syn的积累可增强导致神经退行性变的其他致病机制。其次,蛋白质聚集体可能是由多种致病机制引起的副产物,聚集体本身不具有致病和保护作用。第三,将有毒的可溶性蛋白质聚集体隔离成不溶形式可能是一种神经保护机制方式,可以避免神经元和突触功能障碍,从而延缓神经退行性变过程〔67〕。Yang等〔68〕研究了Parkin和α-syn之间可能的功能相互作用,发现Parkin的共表达导致A53T衰减,A30P突变α-syn毒性减弱。野生型或A30P突变α-syn诱导的黑腹果蝇攀爬能力丧失可以通过Parkin的共表达来抑制〔69〕。在一项使用过度表达A30P突变α-syn转基因Parkin缺失小鼠的研究中发现,Parkin缺乏时运动障碍延迟,含有pSer129的神经元比例降低〔70〕。研究发现,在rAAV载体介导的人类野生型α-syn过度表达后,SNpc中多巴胺能神经元的丢失和pSer129α-syn比例的增加(总α-syn量没有变化)没有差异〔71〕。使用rAAV载体介导的人类野生型α-syn和Parkin在猕猴中的过度表达也获得了类似的结果。Parkin的共表达导致α-syn聚集减弱,pSer129α-syn数量减少。研究表明,Parkin过度表达或Parkin敲除的大鼠嗜铬细胞瘤细胞,减弱了细胞外α-syn低聚物诱导的毒性〔72〕。尽管PARK2患者的尸检神经病理学检查对于路易小体的病理存在尚不明确,但Parkin和α-syn之间功能性作用的发现高度一致。Parkin可以调节α-syn积累、PTM介导的聚集及减弱α-syn介导的毒性方面等功能,那么可以假设α-syn聚集导致细胞内微聚集体的形成可能是PARK2介导的PD的一种潜在细胞内致病机制,该机制比路易小体的形成更为常见。
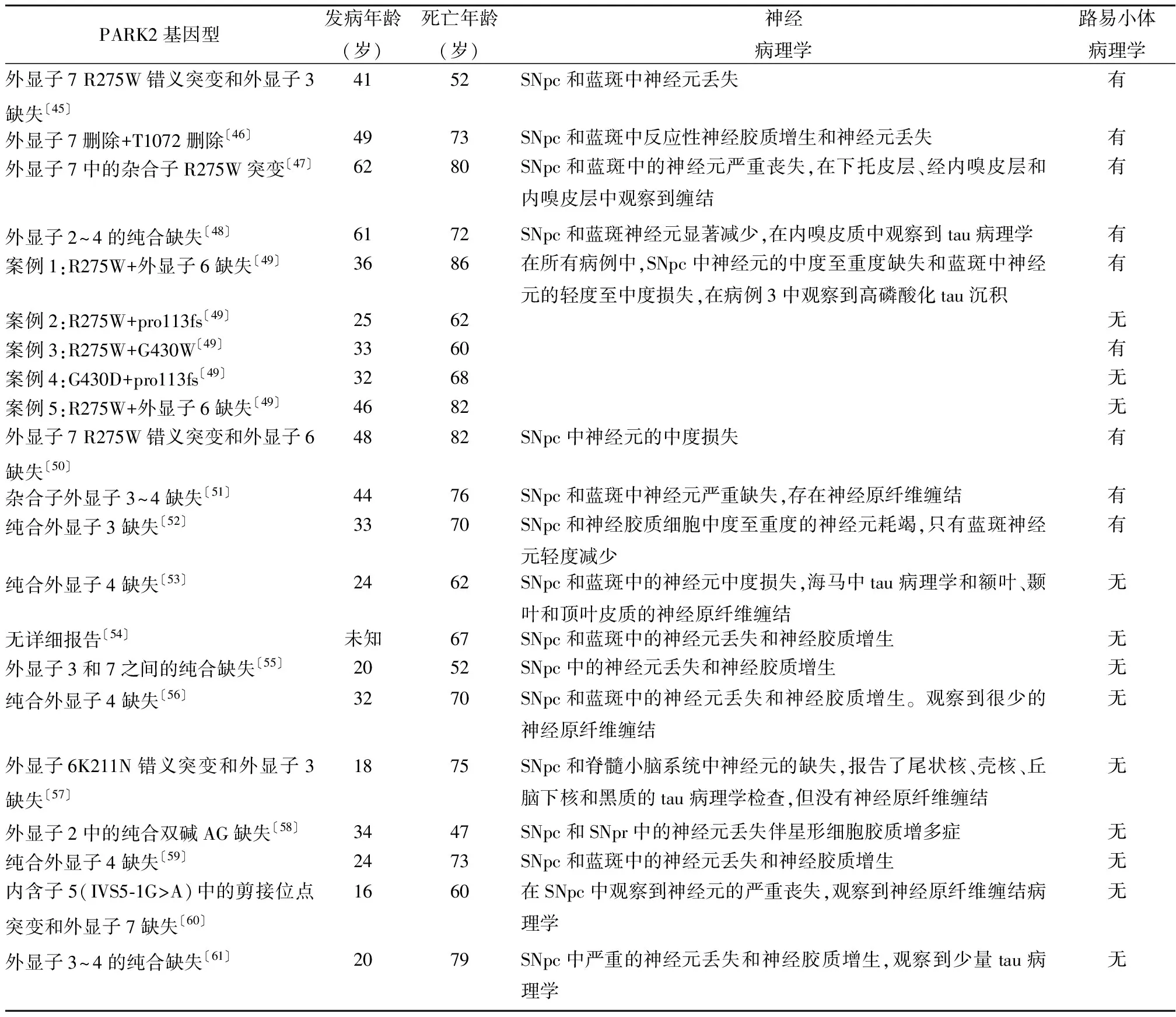
表1 PARK2突变的患者的尸检神经病理学结果
3.4Parkin影响α-syn对细胞凋亡的调控 研究发现,PD患者的SNpc中神经元丢失时,胱天蛋白酶(caspase)3水平升高,Parkin可以通过抑癌基因(p53)依赖性方式阻止caspase3的活化,导致p53 mRNA和蛋白质水平降低〔73〕。因此Parkin作为p53转录抑制因子发挥作用,功能丧失的Parkin突变增加了p53表达,促进凋亡发生。在生理条件下,α-syn会下调p53,抑制细胞凋亡,然而在PD中α-syn聚集导致其无法抑制p53,这将增强p53表达,并导致细胞凋亡〔74〕。
α-syn的过表达以剂量依赖的方式导致核因子(NF)-κB活化减弱,下调抗凋亡因子B细胞淋巴瘤(Bcl)-2表达,上调GSK3β水平〔75〕。GSK3β通过其调节抗凋亡蛋白Bcl-2、髓系白血病(Mcl)-1及促凋亡蛋白(Bax)来促进细胞凋亡〔76〕。GSK3β激活已被证明可以在Ser163处上调和磷酸化Bax,这有助于其线粒体定位〔77,78〕。此外,GSK3β在Ser159处磷酸化Mcl-1,导致其泛素化和降解〔79〕。病理状态下,α-syn介导的GSK3β激活导致Bax介导的线粒体膜孔形成增加,并隔离Bcl-2,从而导致细胞色素c释放到细胞质中。进入细胞质后,与Apaf-1结合激活前蛋白酶9,进而激活下游caspase,启动凋亡过程〔80〕。研究发现,Parkin能够降低GSK3β的表达和活性〔81,82〕,由此发现,GSK3β在调节蛋白质聚集和细胞凋亡方面起着关键作用,而这种机制可能是PD患者Parkin功能丧失的核心。因此,PARK2介导的PD中Parkin功能的丧失不仅可能促进蛋白质聚集,而且可能导致细胞凋亡并介导细胞死亡。由上可知,Parkin对α-syn微聚体的形成具有促进作用,所以,当Parkin发生功能障碍时α-syn聚集,导致细胞凋亡的发生。
综上所述,PARK2介导的PD分子机制是复杂的,需要更多的研究来了解功能失调的细胞内信号传导过程。Parkin功能的丧失可能导致持续形成有毒物质α-syn低聚物和原纤维,破坏许多细胞内信号传导过程,导致细胞凋亡的发生。Parkin可以防止α-syn发生硝化、寡聚化和氧化应激,这对研究如何通过调节Parkin的功能去更好对抗α-syn毒性和tau过度磷酸化以治疗PD具有重要意义。本文介绍了PARK2介导的PD中微聚体的可能,它们导致多巴胺能神经元的严重丧失,但其中所观察到路易体并不一定是致命效应的中介,可能仅仅是病理过程的产物。PARK2介导的PD中病理性细胞内信号传导过程中Parkin与α-syn相关的功能失调途径的共同点可能是蛋白质清除系统受损和蛋白质PTM失衡导致蛋白质错误折叠和聚集。这对研究PARK2介导的PD具有重要意义。
