杂多酸型离子液体杂化材料的第一性原理研究
2024-01-18李丽华
宋 悦,李丽华,王 鹏,吴 限,马 诚
(1.辽宁石油化工大学 石油化工学院,抚顺 113000; 2.中国石油天然气股份有限公司广东石化分公司,揭阳 522000)
1 引 言
杂多酸是一类重要的多金属含氧酸盐材料,由于其优良的性质被广泛用于催化、医学、电化学等多个领域[1-3].但杂多酸稳定性差、易溶于极性溶剂、易潮解等问题阻碍了它的应用.有机-无机杂化方法是改善杂多酸性质的有效且新颖的途径[4].目前报道的以杂多酸作为阴离子,有机聚合物作为阳离子[5],此类杂化物普遍具有较强的酸性可用于工业催化领域中,但较高的成本使之难以推广.而在电化学领域,吴庆银等人以离子液体作为阳离子,杂多酸作为阴离子复合制备出兼具两者优点的新型杂化材料,改善了杂多酸易潮解、离子液体易泄露等问题,拓展了两者的应用范围[6].但目前此类研究仍处于尝试与摸索阶段,目前的研究多是将不同的杂多酸和离子液体进行组合、测试[7].其研究层次也只局限于讨论杂多酸的原子组成或离子液体的烷基链长度等方面对电导率的影响[8],后续研究仍需大量实验去验证,并没有对该方向提供足够的理论依据和指导性建议.不利于杂多酸及其复合材料的长远发展,同时也凸显了目前杂多酸方面的理论发展与实验室研究严重脱节的窘境.
由于计算机科学的发展,可以利用例如密度泛函理论(DFT)[9]等进行量化计算,克服了以前半经验方法对杂多酸体系庞大、复杂而带来的精度低等问题[10].但是目前有关杂多酸的量化计算研究多集中在磁性领域[11],而有关杂多酸及其复合物电化学性质的研究仍为空白.本文首次采用第一性原理计算方法对本文合成的产物进行研究,探究微观结构的差异,并对它们的结合能和载流子有效质量展开模拟分析,计算结果与实验结果吻合.因此本文所提出的模拟计算方法可为以后合成此类新型杂化材料提供新的模拟分析思路和理论依据.
2 实验部分
2.1 三种杂化材料的合成
采用环氧氯丙烷与亚硫酸氢钠作为磺化剂中间体来与1-丙基咪唑反应合成出新型离子液体来改善目前的制备方法.实验分别合成了三种不同钼原子数取代的磷钨酸(H3PW11MoO40、H3PW9Mo3O40和H3PW6Mo6O40)作为阴离子,再与前面阳离子杂化并最终制得[PIM-CHP]3PW11MoO40、[PIM-CHP]3PW9Mo3O40和[PIM-CHP]3PW6Mo6O40三种杂化材料.
2.2 计算工具及参数选择
本文在基于DFT理论框架下,使用Material Studio中的CASTEP[12]模块进行一系列的运算.电子交换-关联能交换势选择了局域密度近似泛函LDA,赝势函数选用CA-PZ,以超软赝势usp法构造赝势[13].能量计算采用的是SCF(自洽迭代法)形式[14],设置的收敛值为0.2*10-4eV/atom.构型优化的截断能(Cutoff Energy)为260.000 eV.第一布里渊区k空间[15],网格点使用Monkhost-Pack方案,设置为2*2*2.采用BFGS法来寻找最稳定构型[16].每个原子在进行运算时,体系的总能量的收敛值取5.0*10-5eV/atom,施加在原子上的最大力为0.1 eV/Å,原子所受的最大应力为0.2 GPa,最大原子位移为0.005 Å.所有优化均在最低能量对称性P1下进行.
3 结果与讨论
3.1 表征测试
对杂化物进行了电导率测试,测试结果如图2所示.三种杂化物均表现出较高的电导率(室温下超过1×10-3S·cm-1),且遵循[PIM-CHP]3PW11MoO40<[PIM-CHP]3PW6Mo6O40<[PIM-CHP]3PW9Mo3O40的关系,三个杂化物均属于新型高电导率固体电解质.
3.2 结构优化
首先对构造的产物进行结构优化,寻找其势能最低构像,解决分子间不合理的高能构型问题.本文中离子杂多酸是对CSCD晶体数据库中的标准12-磷钨酸及咪唑分子进行原子替换和单独结构优化获得.最后利用建模工具将阴阳离子杂化获得三种杂化物的最终稳定构型.本文选用了Castep模块里的BFGS法进行优化[17].优化结构如图1所示.

图1 杂化物的电导率随温度变化曲线
从优化数据来看,随着钼原子取代数的增多,三种杂多酸的P-Oa键长逐渐缩短.但三个杂化物的P-Oa键长明显短于其相应的阴离子杂多酸.且六个钼原子配位的杂多酸及其杂化物会产生轻微形变,这与互相毗邻的钼原子与中心磷原子产生吸引力并更易导致不规则角的非线性结构形成有关,而这种形变会使结构趋于更稳定化.另外三种杂化物的端氧键也相应缩短,这是由于阳离子的插入会对整体结构的对称性产生较大影响.另外IL-PW6Mo6杂化物的微观结构与前两者明显不同,阴阳离子处于高度分散状态,这可能与过量的钼取代数影响原来磷钨酸的结构有关.
3.3 结合能分析
结合能是无相互作用的两体系间聚合的能量,可直接反映同一体系内各组成部分结合的紧密程度和衡量形成化学键的结合强度[18].本文采用软件计算比以往报道中以配位原子和中心原子的种类和数目去描述杂多酸稳定性的结论更具说服力.E表示结合整体所需要的内能,当E为负值时表示两者相互作用可使体系能量降低,而绝对值越大代表体系越稳定.本节所需要求的结合能遵循公式(1):
Ebind=Etotal-Ehetropolyacid-nEinoic-liquid
(1)
式中Ebind为所要求解的结合能,Etotal、Ehetropolyacid和Einoic-liquid分别为模拟计算出的杂化物总能量、阴离子杂多酸的能量及阳离子离子液体的能量.n表示杂多阴离子与阳离子的配位数.各组分内能计算结果如表1所示.

表1 各组分内能数据
将收集到的结果一一对应地放到公式中,计算出三种杂多酸型离子液体地结合能见表2.

表2 各组分结合能
由数据可得IL-PW6Mo6有最稳定的结构.这可能与PW6Mo6阴离子与离子液体阳离子组成了离子型杂化物有关,从模拟优化后的构型来看,杂多酸与离子液体并没有形成共连的“多金属含氧桥”结构,仍以分散的离子对存在,导致了组分间的相互作用力最强.
3.4 导带、价带载流子有效质量
导电本质是材料中带点粒子在电场作用下的定向移动.在导体、半导体材料中导带电子和价带空穴(又称载流子)起主要作用[19].载流子有效质量较小表明其具有较高的迁移率,电导率也较大[20].直接测量载流子的有效质量的方法主要有回旋共振法、Shubnikovde Hass法和磁光效应法.这类方法均对仪器设备、检测环境、样品纯度有较高要求.此外,目前没有关于此类杂化材料的电化学性质的量化计算研究,因此本文首次提出一种新的方法计算并预测出材料的载流子有效质量,从而弥补当前研究领域的空白.并为以后研究此类杂化物提出理论依据.
3.4.1载流子有效质量计算原理
首先将晶体中有效质量张量公式简化为:
(2)
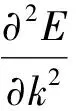
(3)
其中c为二阶函数拟合后的c值,m0为电子静止质量.分析有效质量时通常用所求质量m*与电子静止质量m0的比值来比较载流子有效质量的大小.
3.4.2载流子有效质量计算结果
价带空穴与导带电子计算的载流子有效质量图分别如图2所示.
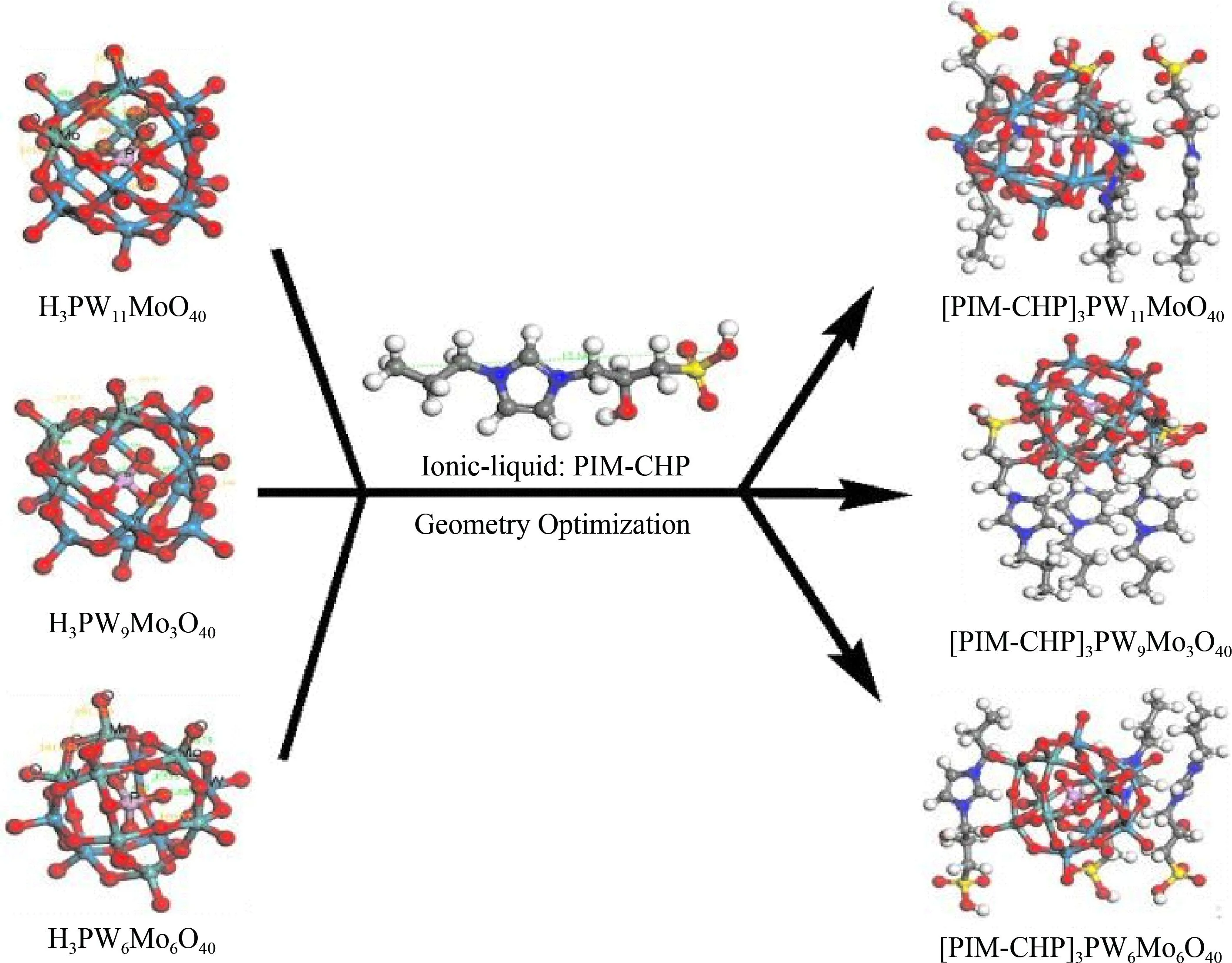
图2 杂化物的稳定微观结构
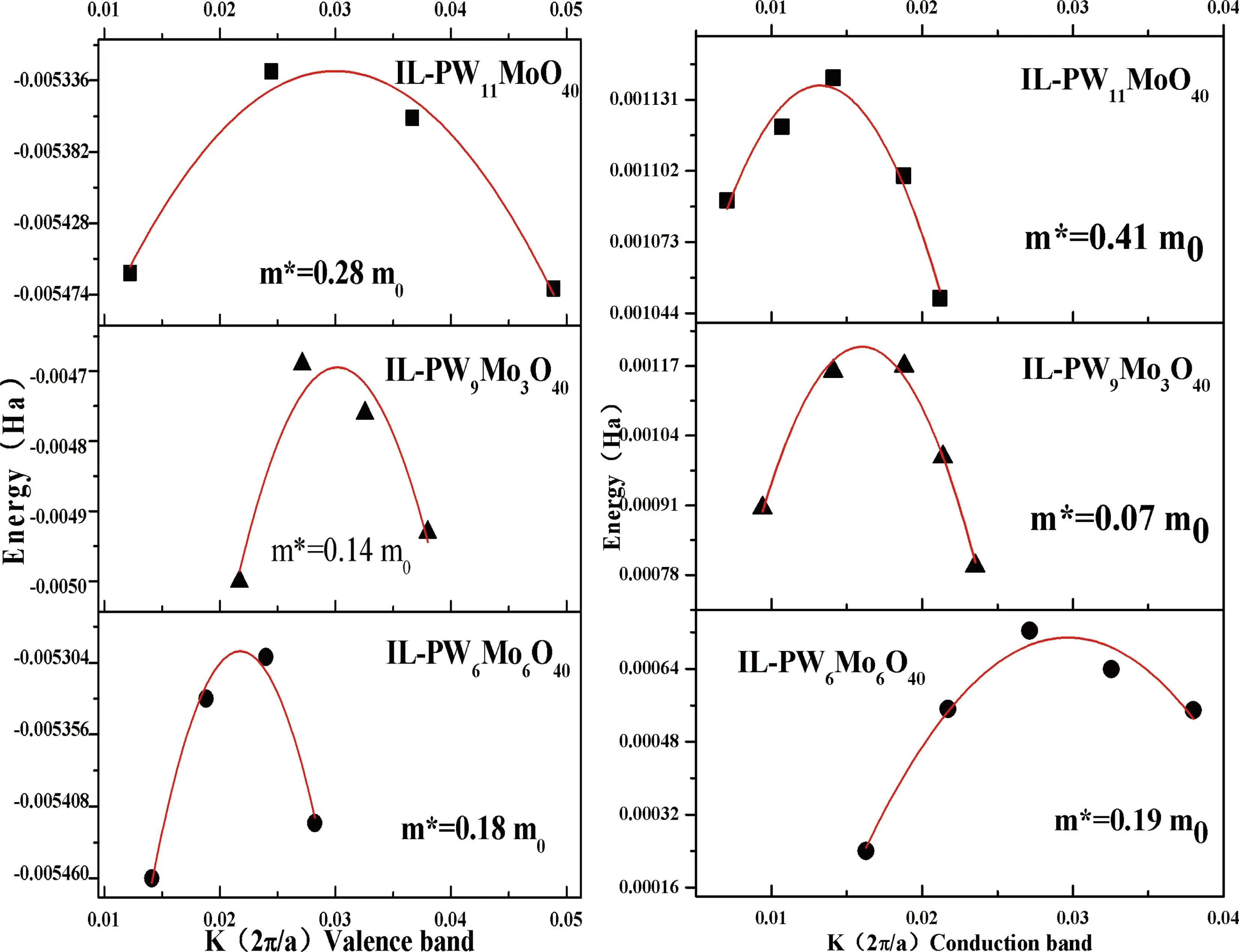
图3 三种杂化物的价带空穴及导带电子有效质量的计算(左:价带,右:导带)
价带空穴载流子有效质量拟合方程分别为(m*:有效质量,m0:电子静止质量):
(IL-PW11MoO40)y=-0.00569+0.02406x-0.4018x2m*=0.28m0
(IL-PW9Mo3O40)y=-0.0084+0.244x-4.04x2m*=0.14m0
(IL-PW6Mo6O40)y=-0.006 + 0.121x-2.774x2m*=0.18m0
导带电子载流子有效质量拟合方程分别为:
(IL-PW11MoO40)y= 9.232E-4+ 0.032x-1.213x2m*=0.41m0
(IL-PW9Mo3O40)y=-6.155E-4+0.228x-7.13x2m*=0.07m0
(IL-PW6Mo6O40)y=-0.0015 + 0.153x-2.575x2m*=0.19m0
整理数据于表3.

表3 价带空穴和导带电子载流子有效质量的数据
从模拟分析数据可看出,IL-PW9Mo3具有最低的有效质量,从而其离子迁移时所受阻力最小.且三种杂化物的载流子有效质量和价带空穴的有效质量遵循:IL-PW9Mo3 针对目前研究杂多酸型离子液体杂化材料量化计算领域的空白,本文首次采用MS软件对一系列的杂化物进行微观结构、结合能、载流子有效质量的模拟分析.本文发现杂多酸的取代配位原子数目会对杂化物的性质产生较大影响,在钼取代的磷钨酸中,当钼为低取代数目时,杂化物具有较低的载流子有效质量,从而表现出较强的导电性.但高钼取代数杂化物的载流子的相对质量较大,因此传输阻力也会相应增大,从而其导电性会弱于低钼取代数目的杂化物.本文所介绍的模拟计算方法可作为后续研究此类杂化材料的预测手段,或为材料的性质分析提供理论依据,从而使该类杂化材料可以尽早地为社会服务.4 结 论
