压力对有机半导体均苯四甲酸晶体结构转变和电子性质的影响
2024-01-18龚智诚陈丽敏刘春生
龚智诚,陈丽敏,张 超,刘春生
(南京邮电大学 先进电磁信息材料与器件研究中心,南京 210023)
1 引 言
作为一种研究新材料、发现新规律的重要手段,高压技术被广泛应用于实验研究中.例如,Drozdov等[1]在155 GPa压强和203 K温度的环境下实现了三氢化硫(H3S)材料的室温超导转变;李洪岩等[2]通过掺入不同含量硼铁的铁基合金,以石墨为碳源,在高温高压条件下合成了含硼金刚石单晶体;赵帅等[3]利用高压手段对二维手性钙钛矿材料光学性质进行了调控.高压状态下,能够有效的缩短原子距离,增加相邻轨道的重叠从而改变晶体的结构和性质.这些研究对探索新材料在高压下的电子性质、平衡态以及光学性质等性质起着至关重要的作用[4].
均苯四甲酸(其化学式为C10H6O8)通常作为一种具有独特的光学和电学性质的有机化合物而受到关注和研究,并在实验和理论方面均取得了一定的进展.其中,基于C10H6O8中不与苯环处于同一平面的羧基更倾向于向不同方向连接金属离子这一特点,研究了4种C10H6O8与Ag盐反应产生的化合物的独特结构、红外光谱、热重分析和发光性能[5];通过改变PH值、反应温度、中心金属和中性配体可以实现C10H6O8配位聚合物结构和组成的多样性,并且为C10H6O8配体的新框架和拓扑结构的设计提供了一个新的例子[6];通过对铽-钆均苯四甲酸络合物的光学性质研究发现Tb-均苯四甲酸能够发射 Tb3+的特征荧光,且钆离子对该体系荧光有明显的增强效应[7].
历年来研究者多是通过对C10H6O8掺杂来改善其光电性质,而通过增加压强对C10H6O8的结构转变、电子和光学性质的研究却很少.受实验技术及条件的影响,高压下晶体的结构和性质仍然是一项具有挑战性的任务.理论计算可以避免这一点,并在较大的压强范围内对材料的性质和结构做出合理的预测[8].因此,本文采用密度泛函理论对C10H6O8晶体结构进行计算[9-11],分析了0-300 GPa静水压作用下C10H6O8的晶体结构、电子性质和光学性质,为研究压强对晶体结构和物理性质的调节作用以及改进有机半导体材料的应用提供了一定的理论参考.
2 计算和结果讨论
本文基于离散傅立叶变换的第一性原理[12,13],通过CASTEP程序[14]ultrasoft pseudopotential赝势方法来近似描述离子和电子的相互作用,广义梯度近似[15]下的PBE-TS泛函[16]用于处理体系的交换关联势能.通过Broyden-Fletcher-Goldfarb-Shanno[17]算法对晶体结构充分弛豫.优化参数为:原子间相互作用力的收敛标准为0.05 eV/Å,单原子能量的收敛标准为1 × 10-5eV,晶体内应力收敛标准为0.03 GPa,原子最大位移收敛标准为0.001 Å,程序对4个参数同时进行优化且均达到收敛标准.晶体中各原子的价电子组态为 C-2s22p2,H-1s1和 O-2s22p4.
根据Barrio等[18]的实验研究,C10H6O8属于三斜晶系,空间群为P1,每个晶胞中含有2个C10H6O8分子(分为P1,P2),其中实验晶胞长度为a= 9.52409 Å,b= 8.48355 Å,c= 7.07606 Å,当给C10H6O8施加0-300 GPa 的静水压力后,晶体的空间群和晶系都不发生变化.晶体晶胞结构和分子模型如图1所示.
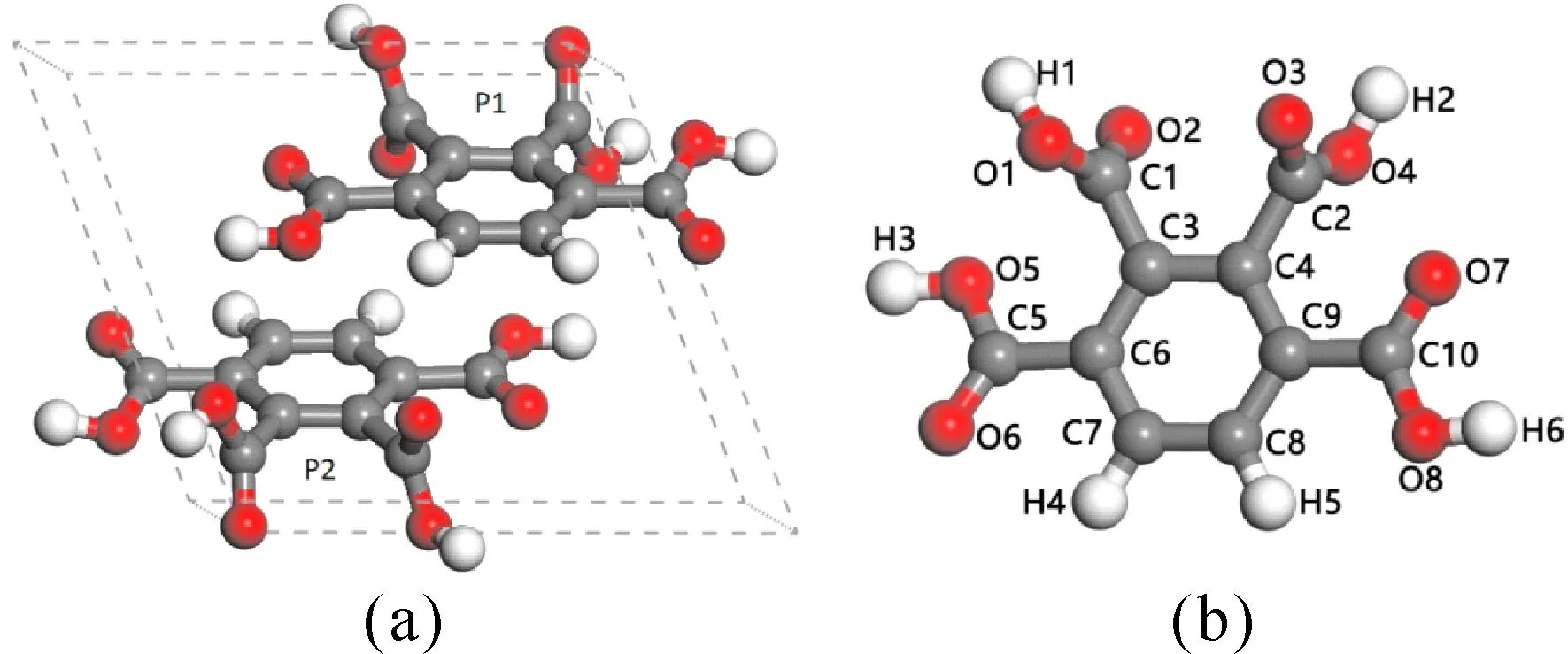
图1 (a)C10H6O8的晶胞,(b)C10H6O8的分子序号
通过晶格参数的测定,不仅可以了解分子晶体的几何结构,也可以由此来判定所采用的理论方法对分子间相互作用描述的可靠程度.初始的C10H6O8晶胞运用PBE-TS 泛函修正方法进行充分优化,所得到的数据[18]记录在表1中并与实验数据进行比较.其中晶格参数a,b,c的误差分别是+0.04%,+0.8%,-0.28%,而晶胞体积的误差是+1.17%.从计算结果来看,PBE-TS方法与实验数据非常接近,因此采用PBE-TS方法开展后续研究.

表1 实验数据与GGA/PBE-TS优化晶格常数的对比
2.1 晶格结构
从图2可知,施加压强后,晶体参数在0-150 GPa范围内总体呈现下降趋势,其中a轴和b轴分别在20 GPa和30 GPa出现过短暂的增加,随后逐渐减少,类似的现象在Li等[19]的研究中也曾被提及,这可能是晶格畸变导致的.同时,有机分子晶体中氢键的存在对晶体的分子构型产生一定的影响,分别对应于该压强下晶体中键O1-H1和O5-H3断开.压强在150 GPa时晶格参数同时出现突变,且b轴与a轴和c轴的增减趋势相反,此时晶体结构可能出现巨大的结构相变.从体积变化中可以看出体积随压强的增加而逐渐减小,与晶格参数相比变化并不明显.因此需要进一步分析高压下 C10H6O8晶体结构和性质的变化,下文中分别对C10H6O8晶体的分子结构、电子结构和光学性质随压强的变化进行研究.
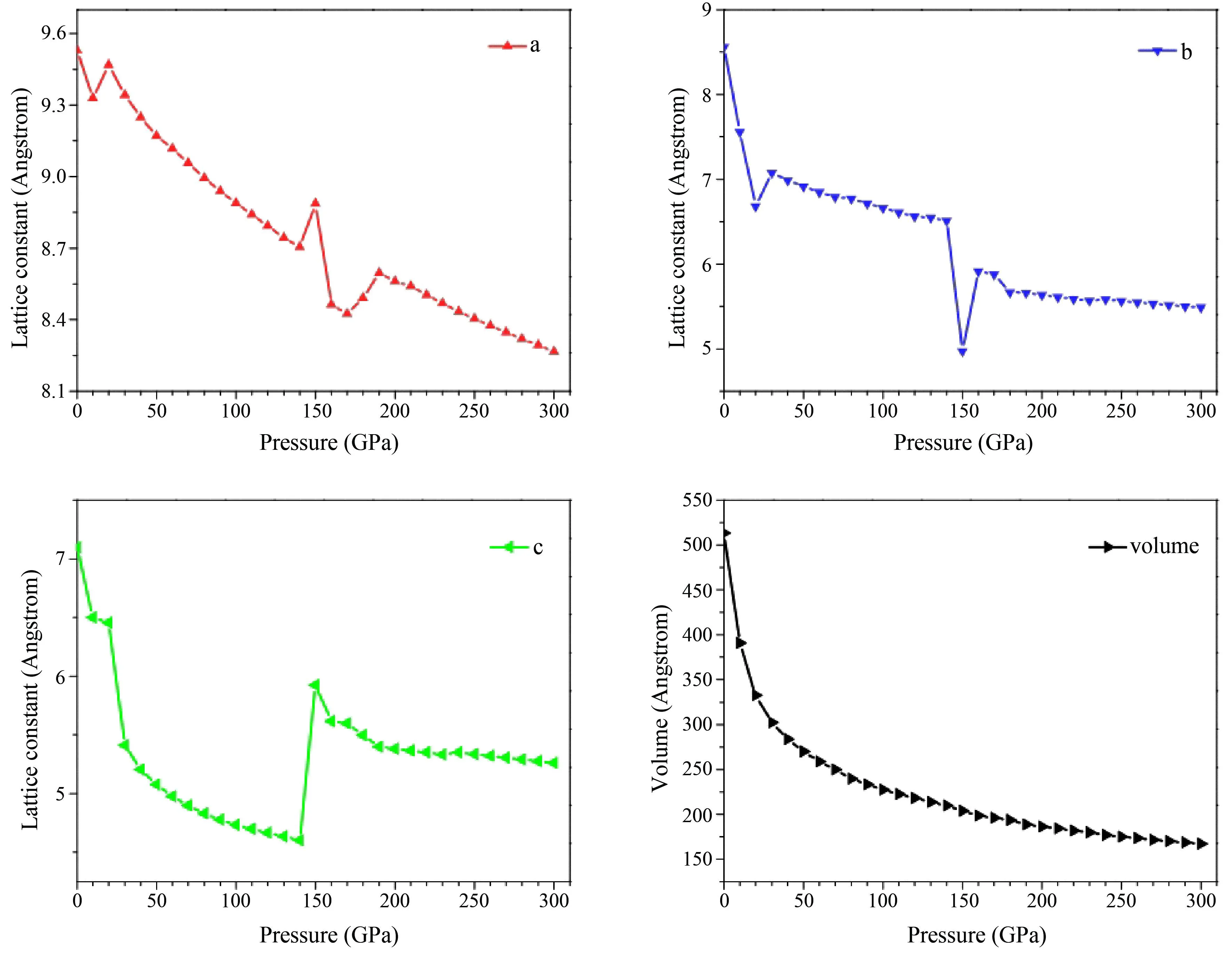
图2 不同压强下C10H6O8的晶格常数(a,b,c)与体积
2.2 分子结构
图3显示了P1和P2分子随压强增大的成键和断裂过程(对称成键不表述).在0 GPa时,P1分子与P2分子距离较远,分子结构较为稳定.随着压强增加,两个分子中原本独立的C1原子和O4原子在80 GPa时相互成键,构成了一个C1-C2-C3-C4-O4五原子环.在150 GPa时,受到分子间相互作用影响,P1和P2分子间距离减小,C3(P2)-O6(P1)成键,同时两个分子中的C6原子也互相成键.在160 GPa时,O1原子代替了80GPa时五原子环C1-C2-C3-C4-O4中的O4原子,构成了一个新的C1-C2-C3-C4-O1五元子环.同时两分子中O6原子与C3原子断开,转而与C4原子成键,而共价键键C8(P2)-O5(P1)也在此时形成.压强增加到190 GPa时,分子间距离进一步减小,五原子环中的C3(P2)原子与C6(P1)原子形成新的共价键,而五原子环C1-C2-C3-C4-O1也在此时断开.直到240 GPa时,两个分子中一直成键的C5原子和O5原子断键,之后此结构不再受压强增加影响保持到300 GPa.上述现象对应于图2中的晶格变化趋势.
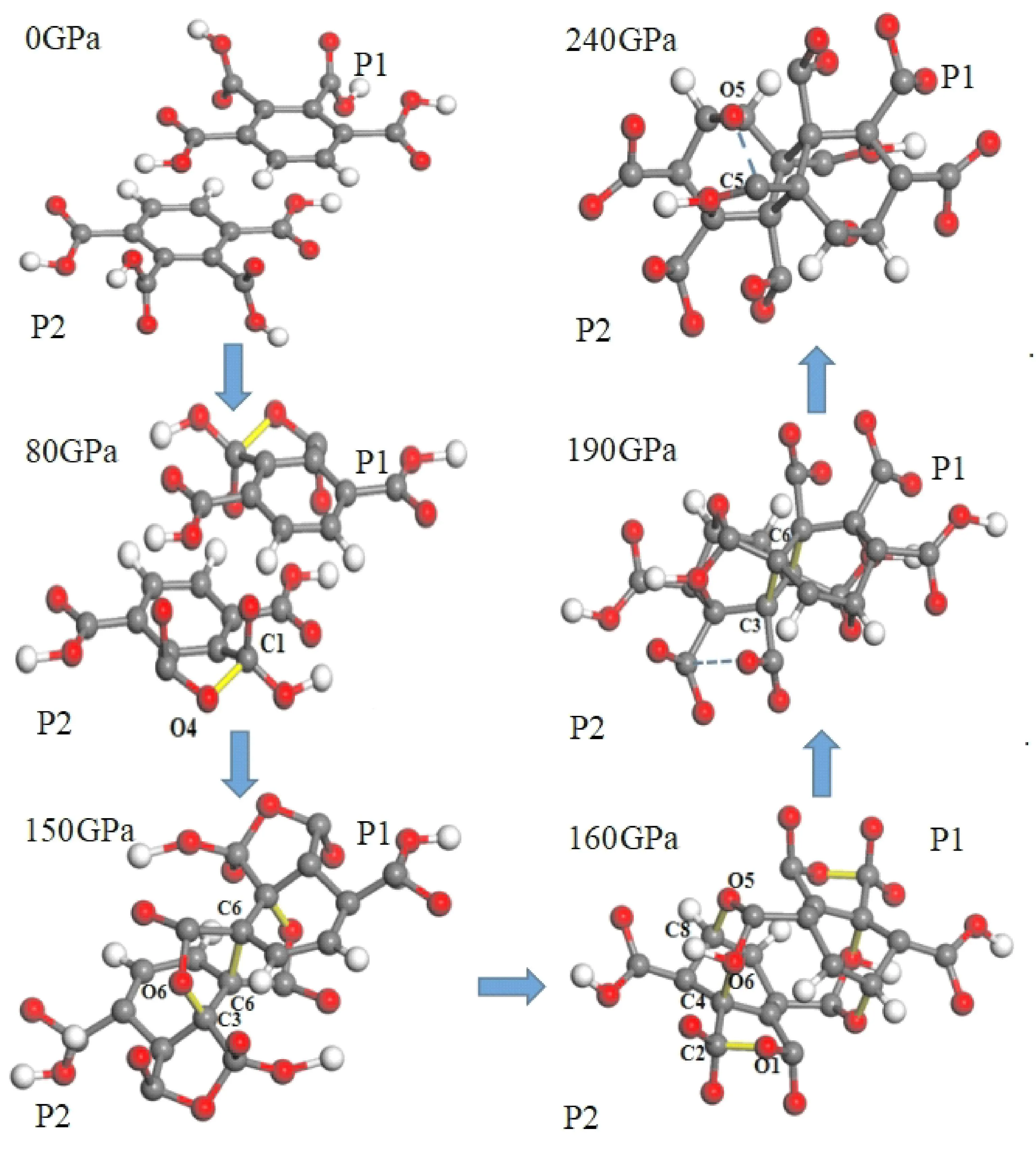
图3 P1和P2分子的成键和断键过程
2.3 电子结构
图4为晶体带隙随压强变化的趋势.随着压强的增加,晶体带隙在0-140 GPa范围内总体呈现下降趋势,之后存在不连续的变化.主要突变点为150 GPa、190 GPa、240 GPa,从上可以看出,带隙突变点和上节的结构突变点基本一致.其中在150 GPa时晶体带隙减小为0,晶体由半导体相转变为金属相,与此对应,晶格参数也发生了明显跳变.
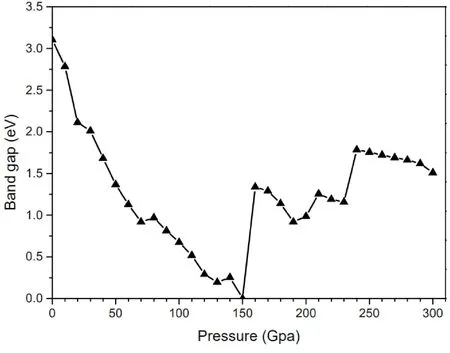
图4 不同压强下C10H6O8晶体的带隙
C10H6O8晶体在0、80、150、160 GPa时的能带结构如图5所示.为了准确观测结果,选择了+ 5 eV到-5 eV的能量范围来分析能带结构.

图5 不同压强下C10H6O8的能带结构
在0 GPa时,它表现为直接带隙半导体,Eg = 3.11 eV.随着压强的增大,晶体带隙逐渐减小,在80 GPa时降至0.619 eV.在这种情况下,导带变宽逐渐向费米能级扩展,价带顶未越过费米能级,晶体仍表现为半导体相,但金属性在压强作用下增强.当压强为150 GPa时,可以发现价带越过费米能级,带隙变为0 eV,表明晶体由半导体向导体转变.此后,晶体由直接带隙转变为间接带隙.
图6为压强为0 GPa和150 GPa时C10H6O8 总态密度和分态密度.显然,C原子和O原子对总态密度的贡献最大,可以推断它们在晶体中具有较高的活性.另外从图中可以看出,原子间的态密度峰有重叠,说明对应的原子间存在轨道杂化,形成了强共价键.在远离费米能级的价带区域,主要由O-2s轨道电子贡献,而靠近费米能级的价带区域则是受C-2p轨道和O-2p轨道电子共同影响.而在导带范围内,主要由C-2p轨道电子贡献.当压强为0 GPa时,在费米能级附近的主要是由C-2p轨道和O-2p轨道的电子共同贡献的,其中O-2p轨道电子占主导低位.而在150 GPa时,态密度峰主要是是由C-2p轨道的电子贡献的,且费米能级附近的态密度峰值较小,此时自由电子增多,晶体结构呈现金属性.

图6 C10H6O8晶体在0 GPa和150 GPa压强下的态密度
2.4 光学性质
介电函数是研究晶体带间跃迁和晶体电子结构的一种有效手段,介电函数的实部通常用于描述材料中电磁场的传播行为,虚部用于描述材料中的光吸收,可以用以下公式描述:
ε(ω)=ε1(ω)+iε2(ω)
(1)
其中ε2(ω)为介电函数的虚部,可根据已占用波函数与未占用波函数之间的动量矩阵元计算.ε1(ω)是介电函数的实部,可以通过虚部从克拉莫-克若尼关系式推导出.
图7为0 GPa时能量范围为0-40 eV内C10H6O8的介电函数实部和虚部.从实部可以看出,常压下静态介电常数ε1(0)=3.5.值得注意的是,ε1在 16.76 eV-21.98 eV之间为负值,表明此时光不能在C10H6O8晶体中传播,该材料呈现出金属反射特性.当光子能量在3 eV附近时,虚部从0开始显著增加.这个临界阈值与0 GPa压强下带隙密切相关.光子能量接近能隙值时,会使价带与导带之间发生光电耦合效应并激发电子跃迁.在3.97 eV时,虚部出现最大峰值,这可能是由于电子从价带顶部直接跃迁到导带底部造成的.随后,在12-18 eV区间形成一个宽峰,由PDOS图看出,这主要是由晶体中C原子轨道的电子跃迁引起的.当能量大于30 eV时,虚部趋于零.

图7 C10H6O8晶体介电函数在0 GPa处的实部和虚部

图8 C10H6O8晶体在0 GPa和150 GPa压强下的电导率实部与虚部
图7 是0 GPa和150 GPa时C10H6O8电导率的实部和虚部.我们知道电导率实部对应着实际的能量耗散[20].表现为150 GPa时实部整体向高能移动,且主峰处幅值增长至约0GPa时的2倍,这是因为压强增加,晶体被压缩,单位体积中导电粒子数增多的缘故.与介电函数虚部对应,0 GPa时电导率的实部峰值在相同的能量附近出现,其中低能峰可能是由于电子跃迁时电子散射引起的.电导率的虚部对应着电场的能量向电子动能的转化,导致电子在外加电场的作用下做简谐振动.在150 GPa时,电导率虚部幅值与0 GPa相比增加更加明显,这意味着驱动电场的能量与电流激发的磁场能量的相互转化变得更加剧烈和频繁.
3 结 论
本文基于密度泛函理论,运用MS软件中CASTEP模块研究了C10H6O8材料的微观机理.采用 PBE-TS 进行对比计算,分析了其结构、电子态密度、能带结构和光学性质,得到了如下结论:
1)在加压过程中,晶格常数发生数次跳变,原子间反复成键/断键,对应晶体结构改变.
2)压强增加后,电子结构中能带展宽,带隙减小,自由电子更容易发生跃迁.当压强为150 GPa时,DOS中价带越过费米能级,带隙变为0,由直接半导体转变为导体,而在160 GPa时又变成了间接半导体.
3)加压至150 GPa后,晶体的电导率实部和虚部均大于0 GPa时的值,这是由于晶体体积压缩,导致单位体积中导电粒子数增多,引起了晶体的电导率增加.
