内源性大麻素2-AG对海人藻酸诱导损伤的大鼠尾状核神经元A型钾通道电流的调制作用*
2024-01-17朱时钰陆永利李自成杨红卫
朱时钰,陆永利,李自成,杨红卫△
[1三峡大学国家中医药管理局中药药理科研三级实验室,湖北 宜昌 443002;2肿瘤微环境与免疫治疗湖北省重点实验室(三峡大学),湖北 宜昌 443002;3三峡大学基础医学院机能学系,湖北 宜昌 443002;4宜昌市三峡中心人民医院神经内科,湖北 宜昌 443002]
内源性大麻素系统由内源性大麻素、与之结合的大麻素受体、负责内源性大麻素合成和降解的酶以及转运体组成,广泛参与人体多种生理或病理生理过程的调节[1]。其中大麻素受体主要包括CB1 受体和CB2 受体:CB1 受体主要分布在中枢神经系统;CB2 受体主要分布在外周免疫系统[1]。2-花生四烯酸甘油(2-arachidonoylglycerol,2-AG)作为人体内含量最丰富的内源性大麻素,是CB1 和CB2 受体的完全激动剂[1-2]。越来越多的证据证明2-AG 可以调节相关离子通道(如钾、钙等离子通道)的电学活性,在神经兴奋毒性损伤中发挥神经保护作用。然而这种保护机制非常复杂,目前尚未被完全阐明[3]。
A 型钾通道是一种电压依赖性钾通道,又称瞬时外向型钾通道,能产生快速激活和失活的瞬时外向钾电流(A-type potassium current,IA),参与动作电位的起始并且是复极化早期外向电流的主要成分,显著调节神经元兴奋性和动作电位形态及复极化[4]。抑制A 型钾通道会导致去极化加速和兴奋性增加;A 型钾通道功能障碍可能导致神经损伤和一些神经系统疾病,如神经性疼痛、帕金森病和癫痫等[5-7]。但其在神经元兴奋性毒性损伤或相关神经系统疾病中的作用机制目前还未完全阐明。如前所述,内源性大麻素2-AG 可以通过调节包括A 型钾通道在内的钾离子通道参与各种机能调节,但其相关机制目前知之甚少。
尾状核(caudate nucleus,CN)是基底神经节的一部分,也是大鼠颅内最大的核团,与大脑的其他部分具有丰富而多样的解剖连接,在认知、学习、记忆和运动等复杂任务的设计和执行中发挥重要作用;其功能障碍见于多种疾病中,包括亨廷顿病、帕金森病和各种形式的痴呆等[8]。
海人藻酸(kainic acid,KA)是从海人藻中提取的兴奋性神经毒性氨基酸类似物,通过激活α-氨基-3-羟基-5-甲基-4-异噁唑丙酸(α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid,AMPA)/KA 受体,导致神经元兴奋性毒性和兴奋性氨基酸毒性作用,诱发神经退行性疾病、癫痫持续状态、学习和记忆障碍等,被广泛用于建立神经毒性和癫痫的动物或细胞模型[9]。因此本研究采用膜片钳技术,观察KA 的兴奋性神经毒性对大鼠原代培养CN 神经元形态和膜上A 型钾通道电学特性的影响;观察2-AG 能否抑制KA 的神经兴奋性毒性作用并对其作用机制进行初步探讨,以期为内源性大麻素治疗相关神经系统疾病提供有效的病理生理学和药理学依据,同时为进一步探究CN 在神经变性疾病中的诊疗作用及机制提供新证据和思路。
材 料 和 方 法
1 实验动物
新生(出生后24 h 内)SD 大鼠,体重5~6 g,雌雄不限,SPF级,由三峡大学实验动物研究中心提供[许可证号为SCXK(鄂)2017-0061]。所有实验规程均遵循三峡大学实验动物伦理委员会制定的标准。
2 主要试剂
DMEM 培养液、Neurobasal A 神经基础培养液、胎牛血清、B27(神经营养因子)和0.25%胰蛋白酶溶液购自Gibco;KA、URB602[一种单酰甘油脂肪酶(monoacylglycerol lipase,MAGL)选择性拮抗剂]、左旋多聚赖氨酸、L-谷氨酰胺、L-谷氨酸、阿糖胞苷、EGTA、4-氨基吡啶(4-aminopyridine,4-AP)、腺苷5'-三磷酸二钠盐(adenosine 5'-triphosphate disodium salt,Na2-ATP)、HEPES、氢氧化铯和氟化铯购自Sigma-Aldrich;2-AG和SR141716(SR1;选择性CB1受体拮抗剂)购自Cayman Chemical;天冬氨酸钾、四乙基氯化铵(tetraethyl ammonium chloride,TEA-Cl)、氯化钠、氯化镁、氯化铯、氢氧化钠、氯化钾、氯化铬、氯化钙和D-葡萄糖均购自国药集团化学试剂有限公司。
3 主要方法
3.1 大鼠CN 神经元原代培养 大鼠CN 神经元提取按照先前的描述进行[10]:(1)取新生(24 h内)SD大鼠数只,于75%乙醇中浸泡消毒3~5 s,断头取脑(尽量保持脑组织完整性,勿破坏皮层),取出脑组织置于盛有DMEM 培养液的培养皿中;(2)在超净台中用眼科镊钝性分离皮层下尾状核,置于另一盛DMEM培养液的皿中,用刀片将尾状核切成小碎块(以上步骤均在冰上操作);(3)将尾状核组织置于含0.25%胰酶和0.02% EDTA 的消化液中(37 ℃)中,用不同管径的巴斯德滴管依次轻柔吹打碎块,使液体浑浊无组织块,于37 ℃中消化15 min,第7 min摇匀一次;(4)消化结束后用与EDTA 等体积的含10%胎牛血清的DMEM 培养液终止消化,混合均匀后,以1 000 r/min离心5 min,弃上清,加神经元原代细胞培养液,缓慢轻柔地吹打细胞,使其呈细胞悬浮液;(5)将细胞悬浮液用200 目滤网过滤,得到滤过的细胞悬浮液,调整细胞密度为(0.5~1)×109L-1;(6)将滤后细胞悬浮液种植于提前用多聚赖氨酸铺板的塑料培养皿(35 mm)中,前后左右晃动培养皿使细胞均匀分布;(7)将含细胞的培养皿置于37 ℃、5% CO2、湿度95%的细胞培养箱内培养。
细胞换液:提取的大鼠CN 神经元培养1 d(24 h内)、3 d 和5 d 用神经元培养液全量换液。第5 天添加阿糖胞苷(终浓度为2.5 mg/L),目的是为了抑制神经胶质细胞生长。加阿糖胞苷24 h后用神经元培养液全量换液。
实验中使用的CN 神经元的培养时间为7~12 d。实验分组为vehicle (VEH)组、KA(10 mg/L)组、2-AG (10 μmol/L)+KA 组、SR1 (10 μmol/L)+2-AG+KA组、URB602 (10 μmol/L)+KA 组和SR1+URB602+KA组。所有试剂按上述分组加入培养液中处理12 h,其中2-AG/URB602+KA 组和SR1+2-AG/URB602+KA组中的KA 在添加2-AG、SR1 和(或)URB602 30 min后加入,SR1、2-AG和(或)URB602同时添加。
3.2 电生理记录 在室温(22~25 ℃)下进行电生理实验。细胞外液冲洗培养液2~3 次,加入氧饱和的细胞外液,在倒置显微镜(IX-71,Olympus)寻找状态良好、膜干净和折光度强的CN 神经元。玻璃电极毛胚(直径1.5 mm;南京六合泉水教学实验器材厂),由P-97 电极拉制仪(Sutter Instrument)多步拉制成玻璃微电极,入水电阻为5~8 MΩ。实验过程中保持电位的钳制及测试脉冲程序的设定、信号采集与储存均由EPC10 膜片钳放大器和计算机运行PATCHMASTER软件(HEKA Elektronik)完成。
IA细胞外液:TEA-Cl 70.0 mmol/L,氯化胆碱70.0 mmol/L,D-葡萄糖10.0 mmol/L,HEPES 10.0 mmol/L,KCl 5.0 mmol/L,CaCl22.0 mmol/L,MgCl2·6H2O 1.0 mmol/L,CdCl20.1 mmol/L。NaOH 调pH 至7.4,4 ℃保存。TEA-Cl 阻断延迟整流性钾离子通道,氯化胆碱阻断钠离子通道,CdCl2阻断电压依赖性钙离子通道。
IA电极内液:K-ASP 118.0 mmol/L,KCl 20.0 mmol/L,EGTA 10.0 mmol/L,HEPES 10.0 mmol/L,Tris-ATP 5.0 mmol/L,MgCl2·6H2O 2.0 mmol/L,CaCl21.0 mmol/L。KOH调pH至7.3,-20 ℃保存。
4 统计学处理
为消除细胞大小的影响,电流值以电流密度(pA/pF)表示。数据和图片由软件SigmaPlot 11.2(Jandel Scientific)和CorelDRAW 9.0 分析处理获得。所有实验数据均用均数±标准误(mean±SEM)表示。采用单因素方差分析进行组间差异的统计学检验,两组间比较采用Student-Newman-Keuls (SNK)-q检验。以P<0.05为差异有统计学意义。
结 果
1 KA 对培养CN 神经元IA密度的影响及2-AG 和URB602对该作用的调制
在全细胞膜片钳记录中,我们首先观察KA 对CN 神经元上A 型钾通道电流的影响。建立全细胞膜片钳记录模式,给予钳制电压-70 mV,施予1 000 ms、阶跃10 mV、-70~+80 mV 的方波脉冲刺激,频率0.2 Hz,在峰值处测量IA的幅值,记录各组IA变化。这些向外电流的最大值被指定为Imax。图1A 是根据上述脉冲刺激记录得到的电流图(来源于各组具有相似膜电容的CN 神经元)。图1B、C 为各种处理因素下IA的电流-电压关系图。(1)原代培养的大鼠CN神经元经KA (10 mg/L)处理12 h 后,观察到从去极化脉冲-20 mV开始,KA诱导的IA密度明显降低。与VEH 组[(162.01±3.32) pA/pF (n=16)]相比,KA 组(n=14)IA密度峰值显著降低到(34.32±2.07) pA/pF(P<0.01),见图1D。这表明KA 显著减弱CN 神经元上的IA。(2)我们前期实验表明,单独使用2-AG 对原代培养大鼠CN 神经元IA无影响[10]。本实验中直接应用2-AG 预处理后(2-AG+KA 组),IA密度峰值回到(113.91±3.29) pA/pF (n=18;P<0.01vsKA 组),见图1D。这表明2-AG 可有效拮抗KA 对CN 神经元上IA的抑制效应。(3)如前所述,2-AG 是CB1 受体的完全激动剂,且CB1 受体在中枢神经系统中广泛表达,因此本实验进一步研究CB1 受体是否参与了2-AG的上述作用。SR1 是CB1 受体拮抗剂,我们前期实验表明单独给予SR1 对原代培养大鼠CN 神经元IA无影响[10]。本实验中给予SR1+2-AG预处理后(SR1+2-AG+KA 组),IA密度峰值降至(43.54±2.67)pA/pF (n=12;P<0.01vs2-AG+KA 组),见图1D。这表明2-AG 拮抗KA 对IA的抑制效应是通过CB1 受体发挥作用的。(4)MAGL是一种参与2-AG生物失活的丝氨酸水解酶,能够水解大脑中约85%的2-AG;抑制MAGL 会提高细胞内2-AG 水平[11]。因此我们使用MAGL 抑制剂URB602 间接上调2-AG 水平。之前本实验室证实单独使用URB602对原代培养大鼠CN神经元IA无影响[10]。在本实验中使用URB602 预处理后(URB602+KA 组),IA密度峰值回到(103.56±3.59) pA/pF (n=17;P<0.01vsKA 组);给予SR1+URB602 预处理(SR1+URB602+KA 组),IA密度峰值降至(43.54±2.67) pA/pF (n=12;P<0.01vsURB602+KA 组),见图1E。这表明,与直接使用2-AG 预处理相似,URB602 拮抗KA 对IA的抑制效应,该效应是通过CB1受体介导的。
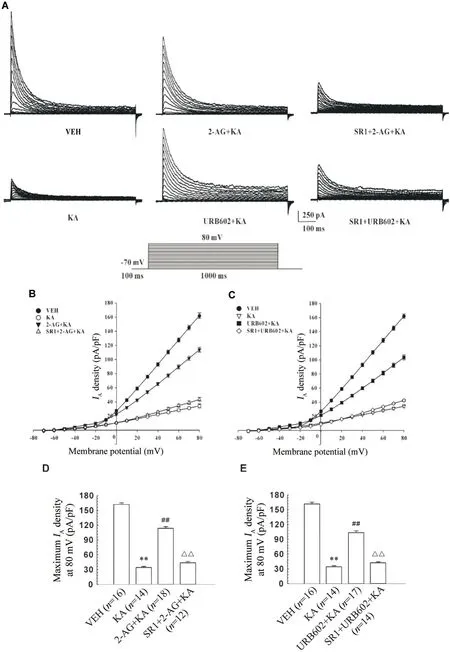
Figure 1. Effects of pretreatment with 2-arachidonoylglycerol (2-AG) or URB602 on the density of A-type potassium channel current(IA) in caudate nucleus (CN) neurons exposed to kainic acid (KA). A: representative IA traces in CN neurons with similar capacitance in vehicle (VEH),KA,2-AG+KA,SR141716 (SR1)+2-AG+KA,URB602+KA and SR1+URB602+KA groups; B: current-voltage relationships of IA in CN neurons treated with VEH,KA,2-AG+KA and SR1+2-AG+KA; C:current-voltage relationships of IA in CN neurons treated with VEH,KA,URB602+KA and SR1+URB602+KA; D: maximum density of IA at +80 mV in CN neurons treated with VEH,KA,2-AG+KA and SR1+2-AG+KA; E: maximum density of IA at +80 mV in CN neurons treated with VEH,KA,URB602+KA and SR1+URB602+KA. Mean±SEM. **P<0.01 vs VEH group; ##P<0.01 vs KA group; △△P<0.01 vs 2-AG+KA or URB602+KA group.图1 2-花生四烯酰甘油或URB602预处理通过CB1受体对海人藻酸损伤尾状核神经元A型钾通道电流密度的影响
2 KA 对培养CN 神经元A 型钾通道激活动力学的影响及2-AG、SR1和URB602对该作用的调节
根据图1 得到的各组电流数据,通过Boltzmann方程I/Imax=1/{1+exp[-(V-V1/2)/k]}拟合得到A型钾通道激活曲线及各组半激活电压(V1/2)和斜率(k),见图2。(1)VEH、KA、2-AG+KA 和SR1+2-AG+KA 组A 型钾通道激活曲线的V1/2值分别为(32.45±2.34) mV(n=16)、(29.10±2.06) mV (n=14)、(31.27±1.98)mV (n=18)和(28.50±1.65) mV (n=12),各组间差异均无统计学意义(图2B);以上4组激活曲线的k值分别 为19.04±1.28 (n=16)、20.19±1.61 (n=14)、20.93±1.89(n=18)和22.84±1.97 (n=12),各组间差异亦均无统计学意义(图2C)。这表明KA 或(和)2-AG 不影响A 型钾通道激活曲线的V1/2值和k值。(2)使用URB602 在KA 处理的CN 神经元中观察到与直接给与2-AG 有相似效果:VEH、KA、URB602+KA 和SR1+URB602+KA 组A 型钾通道激活曲线的V1/2值和k值也均无显著差异(图2E、F),提示使用URB602 内源性上调2-AG,也不影响A 型钾通道激活曲线的V1/2值和k值。以上结果表明,无论是直接应用2-AG 还是通过URB602 提高细胞内2-AG 水平,2-AG的增加都不影响A型钾通道的激活过程。
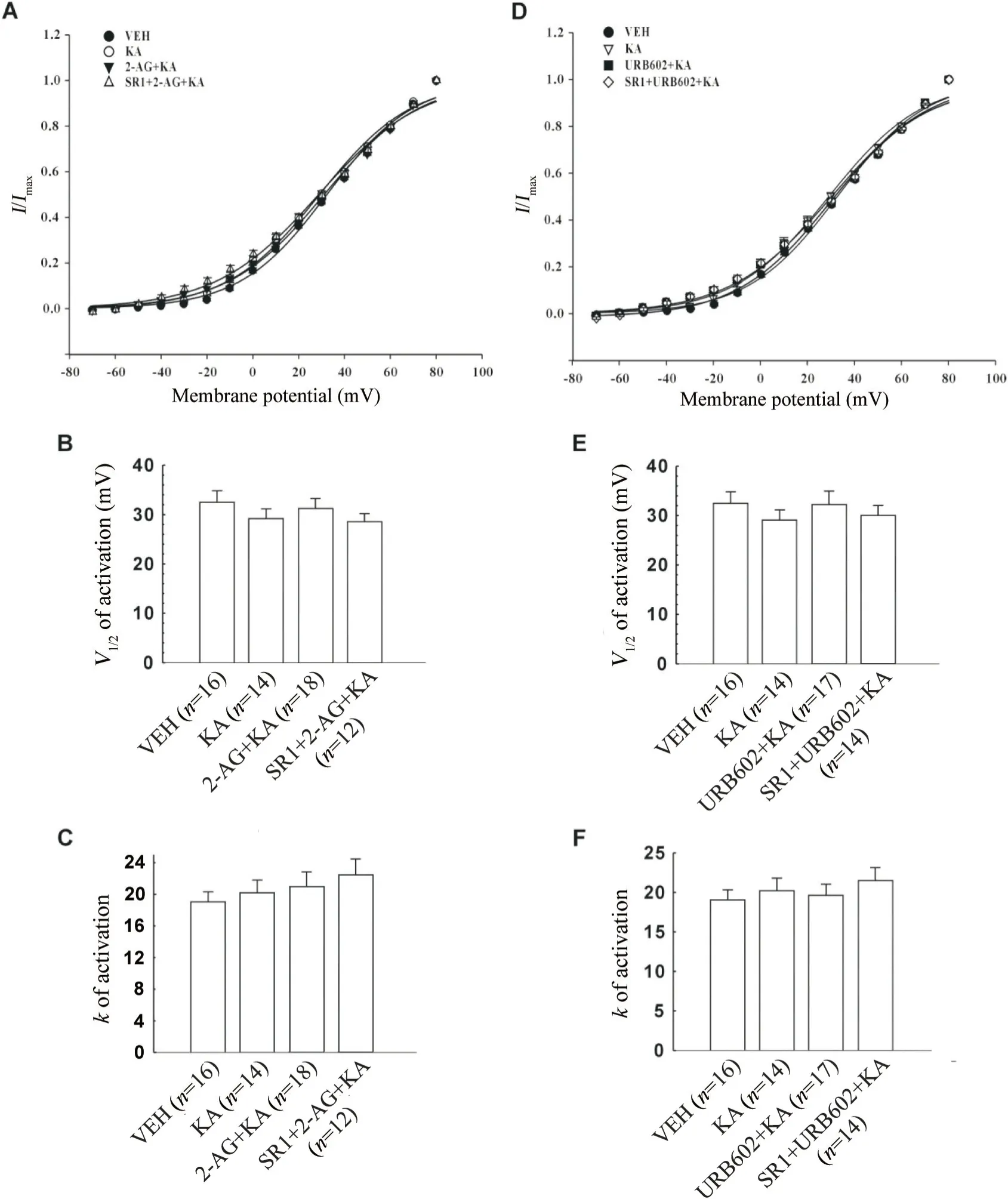
Figure 2. Effects of pretreatment with 2-arachidonoylglycerol (2-AG) or URB602 on activation kinetics of A-type potassium channels in caudate nucleus (CN) neurons exposed to kainic acid (KA). A: steady-state activation curves for A-type potassium channel current(IA) in CN neurons treated with vehicle(VEH),KA,2-AG+KA and SR141716(SR1)+2-AG+KA; B:the semi-activated voltage(V1/2) of IA in CN neurons treated with VEH,KA,2-AG+KA and SR1+2-AG+KA was obtained from the activation curves; C: the slope (k) of IA activation curves in VEH,KA,2-AG+KA and SR1+2-AG+KA groups;D: steady-state activation curves for IA in VEH,KA,URB602+KA and SR1+URB602+KA groups; E: the V1/2 value of IA in CN neurons treated with VEH,KA,URB602+KA and SR1+URB602+KA was obtained from the activation curves; F:the k value of IA activation curves in VEH,KA,URB602+KA and SR1+URB602+KA groups. Mean±SEM.图2 2-花生四烯酰甘油或URB602预处理通过CB1受体对海人藻酸损伤尾核神经元A型钾通道激活动力学的影响
3 KA 对培养CN 神经元A 型钾通道失活动力学的影响及2-AG、SR1和URB602对该作用的调节
A 型钾通道稳态失活曲线由双脉冲刺激法获得:钳制电压-70 mV,条件脉冲为持续时间160 ms、阶跃10 mV、-120~+50 mV 的方波脉冲刺激;每一条件脉冲后紧跟一固定去极化至+50 mV、160 ms 的测试脉冲。根据上述双脉冲刺激记录各组CN 神经元A 型钾通道失活的电流曲线,图3A 显示各组具有相似膜电容CN 神经元的电压-电流原始图。各组电流数据通过Boltzmann 方程I/Imax=1/{1+exp[(V-V1/2)/k]}拟合得到A 型钾通道失活曲线图(图3B、E)及各组半失活电压(V1/2)和k值(图3C、D、F、G)。(1)VEH 组、KA 组、KA+2-AG 组和KA+2-AG+SR1 组的V1/2值分别为(-37.03±3.94) mV(n=16)、(-41.75±4.85) mV(n=14)、(-38.81±3.24) mV (n=18)和(-40.82±3.68) mV (n=12),4 组之间没有显著差异(图3C)。但KA 使失活曲线的k值由11.97±0.95 (n=16)增加到20.81±1.24 (n=14),差异显著(P<0.05)。这些数据表明KA 不影响V1/2值,但增大了失活曲线的k值,提示KA 使A 型钾通道失活的电压敏感性降低[12]。(2)使用2-AG 预处理之后,失活曲线的k值降低至14.20±1.02 (n=18;P<0.05vsKA 组);SR1 和2-AG预处理则将k值提高到17.86±1.37 (n=12;P<0.05vs2-AG+KA 组),见图3D。这提示2-AG 可通过CB1受体抑制KA 对A 型钾通道失活的影响。(3)与2-AG相似,应用URB602 不影响A 型钾通道失活V1/2的均值(图3F),但使失活曲线的k值回降到13.55±0.86(n=17;P<0.05vsKA 组);而SR1+URB602 使k值又回升 到16.34±1.42 (n=14;P<0.05vsURB602+KA组),见图3G。这提示通过URB602 间接增加内源性2-AG 水平,也可通过CB1 受体抑制KA 对A 型钾通道失活的影响。
4 KA 对培养CN 神经元A 型钾通道恢复动力学的影响及2-AG、SR1和URB602对该作用的调节
A 型钾通道恢复曲线的记录:钳制电位-70 mV,施加+50 mV、80 ms的条件脉冲使A 型钾通道完全失活;分别间隔2、4、8、16、32、64、128、256 和512 ms 后再施予第2 次紧接着给予+50 mV、80 ms 的方波刺激(测试脉冲),得到各组A 型钾通道失活后恢复的电流图(图4A)。以测试脉冲电流与峰值电流的比值对时间间隔作图,通过单指数函数I/Imax=1-exp(-t/τ)拟合,其中τ是通道恢复的时间常数,绘制电流恢复曲线(图4B、D)。(1)KA 使τ值从VEH 组的(29.13±2.60) ms(n=14)显著提高到(49.83±4.18) ms(n=11;P<0.01);在2-AG 存 在的情况下,τ值回到(30.90±2.89) ms (n=12;P<0.01vsKA组);此外,当2-AG 和SR1 存在时,τ值显著增加至(48.75±3.67)ms (n=10;P<0.01vs2-AG+KA 组),见图4C。这表明KA 延长了A 型钾通道失活后恢复过程;2-AG 则通过CB1 受体阻止KA 对A 型钾通道恢复过程的影响。(2)与2-AG 类似,当URB602 预处理后,KA 对IA恢复曲时间的影响显著减弱,URB602+KA 组τ值回降到(32.87±3.01) ms (n=13;P<0.01vsKA 组);当KA、URB602 和SR1 联合处理CN 神经元时,URB602的作用被抑制:SR1+URB602+KA 组τ值上升到(46.28±3.86) ms (n=11;P<0.01vsURB602+KA组),见图4E。这表明,通过抑制2-AG的水解增加内源性2-AG 水平也通过CB1 受体显著阻止了KA 诱导的A型钾通道τ值的增加。
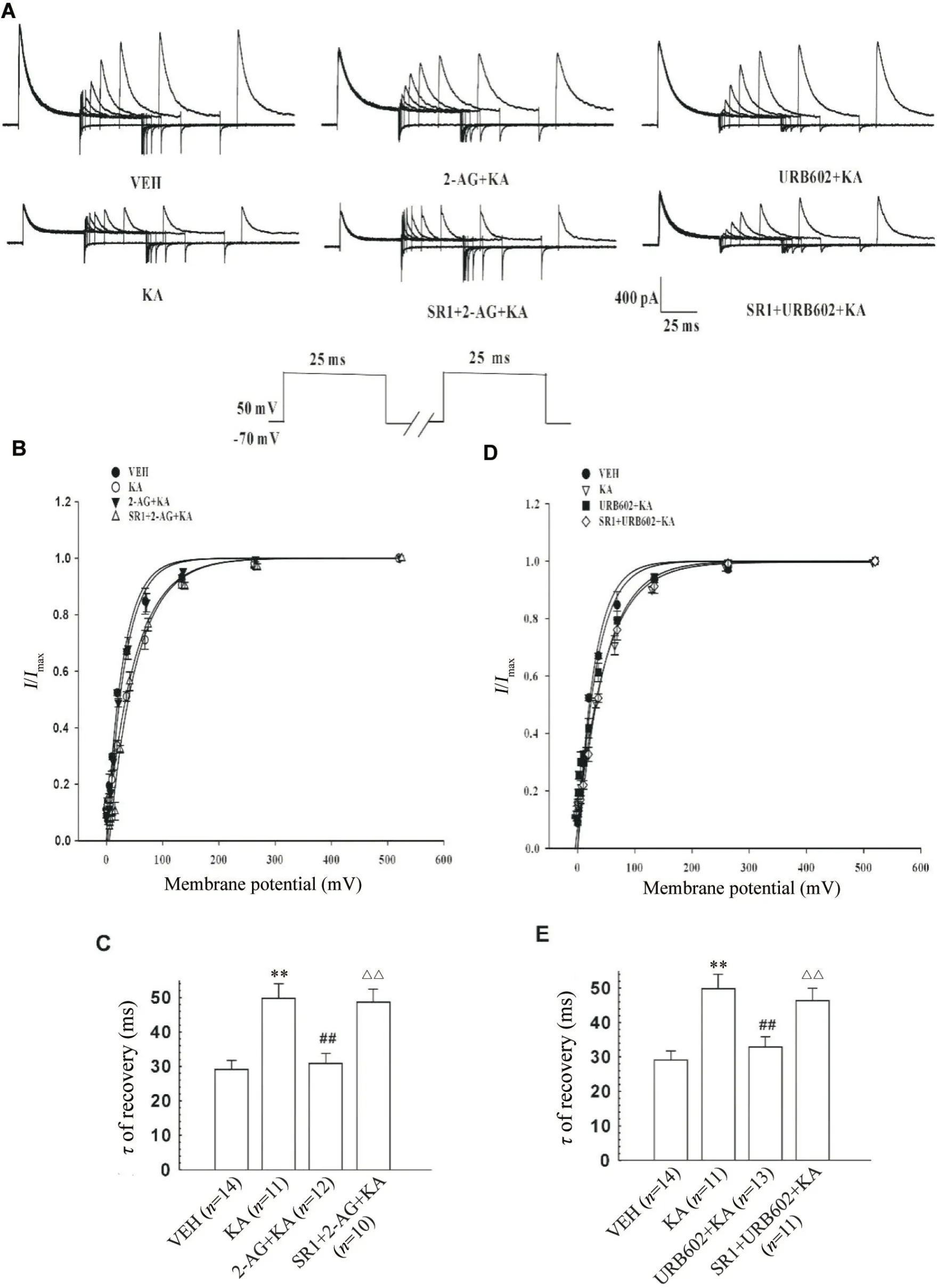
Figure 4. Effects of pretreatment with 2-arachidonoylglycerol (2-AG) or URB602 on recovery kinetics of A-type potassium channels in caudate nucleus (CN) neurons exposed to kainic acid (KA). A: representative A-type potassium channel current(IA)traces in CN neurons under different treatments for the recording recovery kinetics; B: steady-state recovery curves for IA in CN neurons treated with vehicle (VEH),KA,2-AG+KA and SR141716 (SR1)+2-AG+KA; C: the recovery time constant(τ) of IA in CN neurons treated with VEH,KA,2-AG+KA and SR1+2-AG+KA was obtained from the recovery curves; D:steady-state recovery curves for IA in VEH,KA,URB602+KA and SR1+URB602+KA groups; E: the τ value of IA in CN neurons treated with VEH,KA,URB602+KA and SR1+URB602+KA was obtained from the recovery curves. Mean±SEM.**P<0.01 vs VEH group; ##P<0.01 vs KA group; △△P<0.01 vs 2-AG+KA or URB602+KA group.图4 2-花生四烯酰甘油或URB602预处理通过CB1受体对海人藻酸损伤尾核神经元A型钾通道失活后恢复动力学的影响
讨 论
兴奋性毒性是很多神经系统损伤和疾病的重要病理生理机制,用于解释这些神经系统疾病背后的共同的病理基础,如阿尔茨海默病、帕金森病、亨廷顿病、肌萎缩性侧索硬化症、中风和癫痫等[[13]。
作为钾通道超家族中的主要成员,A 型钾通道参与了神经细胞膜电位稳定的维持。当A 型钾通道被启动,细胞膜再次去极化而兴奋时,可以延迟去极化达阈电位的时间[4,13];通道产生的IA是参与神经细胞动作电位复极化早期的主要外向电流,影响动作电位的阈值、形态和时程,是神经元兴奋性的重要决定因素[14]。A 型钾通道功能异常或缺失则会表现出神经元过度兴奋性,导致神经损伤和某些神经系统疾病:例如IA的降低使CA1 神经元的兴奋性增加,导致颞叶癫痫[15];而IA的上调可抑制癫痫小鼠模型中的神经元过度兴奋,并降低了癫痫发作频率[16]。Shinoda 等[17]发现,第5 腰椎脊神经结扎所致神经病理性疼痛模型大鼠α神经纤维IA密度显著降低,表现出更高的去极化静息膜电位和兴奋性;神经营养因子治疗则逆转了IA的减弱,发挥出镇痛作用。因此我们推测KA 所致CN 神经元兴奋性损伤中可能有IA的改变,并通过实验进行了验证。
本实验通过膜片钳实验观察到KA 预处理可以明显降低CN 神经元IA峰值,这与上述IA和神经损伤相关疾病研究的实验报道一致,结合本实验结果提示CN 神经元中IA的抑制与KA 所致的CN 神经元兴奋性毒性之间存在联系。进一步研究表明,KA 对CN 神经元A 型钾通道激活动力学特性和失活V1/2没有影响,但使失活曲线k值增大,失活的电压敏感性降低[12];KA 能显著增大CN 神经元A 型钾通道失活后的τ值,意味着A型钾通道在失活后需要更长的时间才能再次打开[12]。这些结果提示,KA 通过影响A型钾通道的失活和失活后的恢复致使CN 神经元IA减弱。鉴于IA的变化与神经元兴奋性密切相关,IA的减弱可增加细胞兴奋性甚至引起细胞的兴奋性毒性,并最终导致神经元凋亡或死亡[18],因此结合相关文献和本实验数据,我们认为KA 通过影响A 型钾通道失活曲线的k值和τ值等电学活性导致IA密度降低,继而引发神经元细胞兴奋性增高,是KA 造成CN神经元兴奋性毒性损伤的机制之一。
既往实验研究包括我们自己的研究已经表明,内源性大麻素可以调节相关离子通道发挥神经保护作用[19-20],而CB1受体激活也可以打开钾离子通道引起钾的涌入[10,21]。据此我们推测对IA的调节可能是2-AG 抑制KA 诱导的CN 神经元兴奋性毒性的机制之一。本实验通过膜片钳技术记录显示2-AG 显著增加KA 兴奋性神经毒性损伤CN 神经元上IA电流幅度,并有效拮抗KA 所致的A 型钾通道失活后τ值和失活曲线k值的增大,加快了通道失活后恢复过程。随后选取了CB1 受体拮抗剂SR1 为研究药物,实验显示SR1 抵消了2-AG 的上述作用。以上结果表明,2-AG 通过CB1 受体调节CN 神经元A 型钾通道的功能来实现对IA的调制,从而减轻KA 对IA的影响。如前所述,IA的降低可以导致神经元过度兴奋,最终导致神经元的兴奋性毒性[18]。因此,对KA 所致IA减小的拮抗意味着减轻CN 神经元的过度兴奋。以上结果说明2-AG 通过CB1 受体拮抗KA 所致CN 神经元IA的减小可能是2-AG 对抗KA 的神经兴奋性毒性作用、发挥神经保护作用的机制之一。
本实验研究中使用2-AG 降解酶MAGL 抑制剂URB602得到了与内源性大麻素2-AG类似的效果,提示直接增加内源性大麻素2-AG,或者抑制MAGL 使2-AG 水解减少而间接升高细胞内2-AG 水平,在KA所致神经兴奋性细胞模型中同样具有神经保护作用。
内源性大麻素系统是人类机体中一个非常重要的内源性系统,参与了包括情绪调节、疼痛管理、认知功能、奖赏、食欲、脂肪和葡萄糖代谢、神经发生、神经保护、炎症和免疫功能等在内的大量生理过程[22]。因此,以包括2-AG 和MAGL 在内的内源性大麻素系统相关蛋白为目标的化合物已被用于或研究用于疼痛、癫痫、精神疾病、代谢疾病、神经退行性疾病和肿瘤等的治疗[23-24];大麻素相关药物及其机制的研究也因此成为近几十年来的研究热点,也给上述难治性疾病的治疗带来希望。但目前对其作用机制的有限理解限制了该类药物的临床应用。本研究首次提供了KA 的神经毒性作用和2-AG 的神经保护作用与IA直接相关的证据,提示A 型钾通道可能成为治疗兴奋性神经毒性所致神经系统损伤或疾病的有效靶点;也对理解内源性大麻素对人体的调节机制和探索其临床应用途径开拓了新视角。
此外,帕金森病、亨廷顿病和阿尔茨海默病等神经退行性疾病目前尚无有效治疗手段,但越来越多研究证实CN与这些疾病的病理及病理生理机制密切相关:CN 萎缩是亨廷顿病一个已经证实的神经影像学特征[25];研究还发现亨廷顿病患者症状发作前CN 就已经受到显著影响[26];与主观和轻度认知障碍患者相比,阿尔茨海默病患者的尾状核体积更大[27];多数帕金森病患者中存在CN 多巴胺功能障[28];有氧运动诱发CN多巴胺释放增加则与帕金森病患者的运动治疗效果相关[29]。由此可见,CN 在神经系统相关疾病的预防、诊断和治疗中具有重要的研究价值。本实验首次证实CN神经元上A型钾通道参与了2-AG对KA损伤神经元的保护作用,也为上述难治性神经退行性疾病的研究提供了新思路。
