Ⅰ型唾液酸沉积症4例☆
2024-01-08蔡曼斯李洵桦陈子怡罗玥陈定邦
蔡曼斯 李洵桦 陈子怡 罗玥 陈定邦
唾液酸沉积症(sialidosis)是一种罕见的常染色体隐性遗传疾病,由编码唾液酸酶(即N-乙酰神经氨酸酶)的NEU1基因突变所致,主要病理特点是唾液酸寡糖在细胞内的异常蓄积[1-2]。该病最早在1977年由DURAND提出[3],后来Lowden和O’Brien根据起病年龄和疾病的严重程度将其分为I型(轻型)和II型(重型)[2]。Ⅰ型又称黏液脂质病、樱桃红斑肌阵挛综合征,起病较晚,症状表现较轻。Ⅱ型较Ⅰ型多见,起病较早,常表现为Hurler综合征,可见面容丑陋、骨骼异常、肝脾肿大、智力障碍等[2,4]。收集2017年3月至2022年5月就诊于中山大学附属第一医院神经内科4例Ⅰ型唾液酸沉积症患者资料,该4例患者临床表现较为典型,且有3例NEU1基因检测为c.544A>G纯合突变。
1 临床资料
病例1,男,17岁,因“四肢抖动3年余”于2021年7月21日就诊。患者于2017年10月出现双手轻微抖动,紧张或做精细动作时出现,后出现双下肢不自主抖动。症状逐渐加重,伴起步困难,易跌倒。予氯硝安定及美多芭治疗后症状稍缓解。既往史、个人史、家族史无特殊。见图1。查体:神清,高级皮层功能正常。视力粗测正常,未见眼震,构音欠清。四肢肌张力低,肌力5级。双侧指鼻及跟膝胫试验不准。Romberg征阳性。双手平伸可见震颤,行走时可见四肢抖动,冻结步态。四肢腱反射亢进,双侧Rossolimo征及Hoffmann征阳性。颅脑MR平扫(2018年12月22日)未见异常。OCT(2020年8月20日):右眼颞上方视网膜神经纤维层临界变薄。脑电图(2020年8月22日):界限性脑电图(α波调幅欠佳)。NEU1基因检测结果:患者具有纯合突变c.544A>G(p.Ser182Gly),其父母未完善NEU1基因检测。
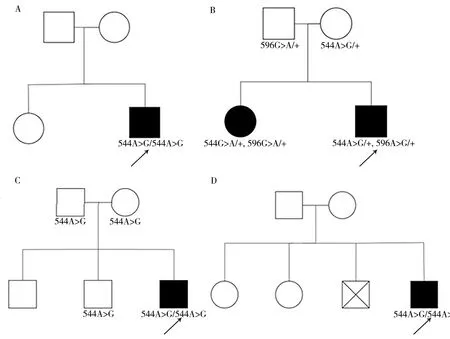
图1 家系图 病例1、3、4 (A,C,D)患者有疾病表征,其父母及兄弟姐妹表型正常。病例2(B)患者及其姐姐有疾病表征,其父母表型正常
病例2,男,42岁,因“反复发作性抽搐30年,四肢抖动20年”于2022年5月20日就诊。患者于1992年(12岁)开始出现反复发作性四肢抽搐,发作时神志不清、口吐白沫,2001年服用德巴金及氯硝安定后未再发作。2002年出现头颈部及四肢不自主抖动;伴行走不稳,易跌倒,伴视物模糊。2018年5月至我院就诊,予妥泰、德巴金、氯硝安定治疗,症状仍缓慢加重。既往史,个人史无特殊。家族史:姐姐有类似症状,已卧床3年,生活无法自理。半年后随访,患者肢体抖动及视物模糊症状较前加重,行走需人搀扶,仅能看见眼前物体。查体:神清,高级皮层功能正常。双眼视力约眼前10 cm指数。可见粗大的垂直眼震,言语稍慢,欠流利。四肢肌张力低,肌力5级。双侧指鼻试验欠准,Romberg征阳性,活动时可见四肢明显抖动。四肢腱反射弱。眼底检查(2018年5月18日,外院)示双侧视网膜樱桃红斑(未找到相应图片)。颅脑MR平扫(2022年4月22日):小脑萎缩,见图2。动态脑电图(2022年10月17日):未见痫样放电。NEU1基因检测结果:患者存在c.544A>G和c.596G>A复合杂合突变,突变分别来自于患者父母,c.596G>A为错义突变,引起编码蛋白质氨基酸的第199位由Gly变为Glu。该突变在国内外为首次报告,在常见的基因突变数据库包括人类基因突变数据库HGMD、Clinvar未有记录,在千人基因组计划中也未查询到。序列对比可见Gly199残基在物种间保持进化保守,可见对蛋白质结构很重要。见图3。使用Polyphen2预测该突变的致病性,评分为0.998(评分越接近于1,损害可能性越大;越接近于0,损害可能性越小),Mutpred2评分为0.910,提示很可能致病,且预测该突变的分子机制为影响跨膜蛋白的结构,综上考虑该突变为致病变异。
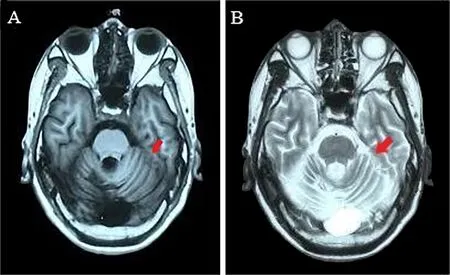
图2 颅脑MR Ⅰ型唾液酸沉积症病例2的颅脑MR中T1WI(A)及T2WI(B)序列可见小脑萎缩

图3 基因序列对比 病例2的NEU1基因突变位点c.596G>A对应的第199位氨基酸在不同物种的比较,黑色方框示p.Gly199位点
病例3,男,27岁,因“四肢不自主抖动8年,视物模糊4年”于2022年5月16日就诊。患者2014年开始出现双手抖动。2018年出现双下肢不自主抖动,伴视物模糊。2020年就诊于我院,予“美多芭、氯硝安定”治疗,服药后起初症状稍缓解,后仍缓慢加重。2022年1月出现言语欠清,伴行走困难,无法独立行走。既往史、个人史、家族史无特殊。半年后随访,患者服药后症状稍好转,可独立行走,视力下降明显,仅能看清1 m内物体。查体:神清,远近记忆力及计算力减退,MMSE量表 23分。视力:OD 0.6,OS 0.5。可见终末眼震,言语欠清。四肢肌张力正常,肌力5级,双侧指鼻及跟膝胫试验不准,Romberg征阳性。活动时可见四肢及颜面部不自主抖动,共济失调步态。四肢腱反射亢进,双侧Rossolimo征(+)。辅助检查(2022年5月16日):眼底镜检查未见异常。视野:双眼生理盲点扩大,大部分视野缺损,光敏感度下降。OCT:左眼颞侧RNFL厚度边界变薄。体感诱发电位结果:刺激双侧上肢正中神经诱发的P15、N20(皮层电位)波幅增高(巨大电位),刺激双侧下肢胫后神经诱发的P40、N50(皮层电位)波幅明显增高(巨大电位)。视觉诱发电位结果:双侧全视野黑白棋盘格翻转视觉诱发电位P100波峰潜伏期延长、波幅低,波形分化重复性差。见图4。脑电图:轻度异常脑电图(安静闭目β活动增多)。颅脑MR平扫:未见异常。NEU1基因检测结果:患者具有纯合错义突变 c.544A>G(p.Ser182Gly)。患者纯合,父母杂合,突变基因分别来自其表型正常的母亲和父亲。

图4 诱发电位结果 病例3体感诱发电位见刺激双侧上肢正中神经诱发的P15、N20(皮层电位)波幅增高(巨大电位)(A,红色箭头处),刺激双侧下肢胫后神经诱发的P40、N50(皮层电位)波幅增高(巨大电位)(B,红色箭头处)。视觉诱发电位见P100波峰潜伏期延长(C)
病例4,男,42岁,因“肢体抖动27年,视物模糊6年”于2017年3月14日就诊。患者1990年出现双上肢不自主抖动。2010年出现双下肢抖动,伴行走不稳;2011年出现视物模糊,2014年出现言语不清。2019年出现抽搐1次,表现为神志不清、四肢抽搐,服用丙戊酸钠及左乙拉西坦后未再发作。既往史、个人史无特殊;家族史:父母为近亲结婚。5年后随访,患者行走不稳加重,走路需人搀扶,视力下降明显。查体:神清,皮层功能正常。双眼视力0.1,未见眼震。爆破性语言。四肢肌张力低,肌力5级。双侧指鼻及跟膝胫试验尚准,Romberg征阴性。双上肢可见意向性震颤,宽基底步态。四肢腱反射亢进,病理征阳性。辅助检查:眼底镜检查(2017年3月14日)未见异常。视野:双眼生理盲点扩大,大部分视野受损。脑电图:轻度异常脑电图(额中线θ活动增多)。颅脑MRI平扫+增强:双侧额叶白质内多发缺血灶。NEU1基因检测结果:患者具有纯合突变c.544A>G(p.Ser182Gly);其父母未进行NEU1基因检测。
2 讨论
Ⅰ型唾液酸沉积症是唾液酸沉积症的一种类型,其机制在于,NEU1编码的唾液酸酶催化糖蛋白、糖脂等糖链末端唾液酸残基的裂解,其功能障碍导致唾液酸寡糖在细胞内的异常蓄积及从尿液中排泄[5],从而导致唾液酸沉积症。该疾病起病时间和症状的严重程度与突变位点以及残存的酶活性相关[6]。
本文报告4例患者均在青少年起病,见表1,病程中都有进行性肌阵挛的症状,表现为发作性肢体抖动,多在紧张、劳累、做精细动作时出现,偶有突发跌倒,与该疾病特征性肌阵挛相一致。本病中肌阵挛主要来源于皮层及皮层下[7],遗憾的是,该4例患者脑电图并未见提示肌阵挛来源的证据,可能与抗癫痫药物的使用有关。4例患者均存在指鼻试验、跟膝胫试验不稳或宽基底步态等共济失调的体征,但颅脑MR检查中,仅病例2见小脑萎缩,病例4在起病27年后颅脑MR仍未见明显异常。国内报告的Ⅰ型唾液酸沉积症患者中,大多数患者MR也未见异常,仅26.7%患者见小脑萎缩。研究指出,病理上许多神经元中可见唾液酸寡糖在细胞质的积累,但即便在起病15年后唾液酸寡糖大量蓄积的情况下,仍没有明显的神经元丢失,这可以解释影像学检查少见异常的原因[8-9]。3例患者存在明显的视力下降表现,仅1例眼底检查见樱桃红斑,樱桃红斑在临床上检测率不高,部分患者早期可能不出现眼底樱桃红斑,也可能在疾病后期消失[10],可见其并非诊断所必需[11]。本组脑电图阳性率较低,国内外相关报告也可看到类似的脑电图结果:国内所报告的病例中有10例完善了脑电图检查,其中有6例结果异常[12-17],多数表现为广泛棘慢波,而国外27例完善脑电图的患者仅有11例结果异常[18],这可能与服用抗癫痫药物后癫痫发作得到控制有关。病例2视觉诱发电位(visual evoked potential,VEP)见P100波峰潜伏期延长,体感诱发电位(somatosensory potential,SSEP)可见双侧上下肢皮层电位波幅增高(巨大电位),这与既往报告一致,SSEP的巨大皮层电位可提示皮层感觉运动整合的过度兴奋[10]。
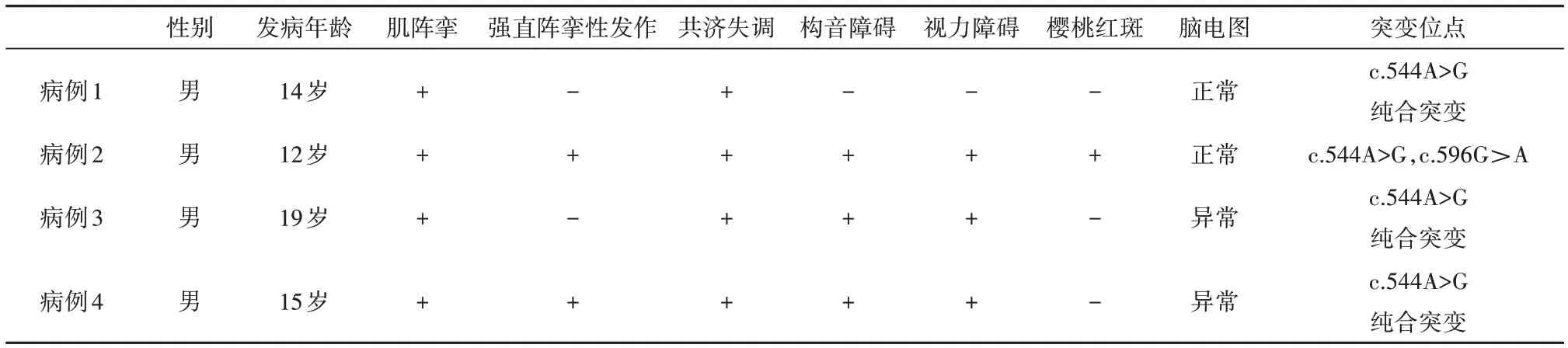
表1 4例唾液酸沉积症患者的临床表现和实验室检查
目前中国大陆报告Ⅰ型患者共有15例[12-17,19-21],分析数据可见中国患者发病年龄较小,平均发病年龄为12岁;大多数患者随着病情进展逐渐出现肌阵挛、共济失调和强直阵挛发作的所有表现。目前国内最常见的突变位点为c.544A>G(80.0%),其次是c.239C>T(53.3%)。本组中,4例患者都存在c.544A>G突变。该突变导致的Ser182Gly突变体位于唾液酸酶的外周,不会明显影响唾液酸酶的分子结构,因此可以表达相对较高的酶活性(约40%的正常的酶活性)[22],这也可以解释,大多数携带该突变基因的患者临床症状相对较轻。
本组症状表现较为典型,起病年龄较晚,存在肌阵挛、共济失调、视力下降等常见表现,无明显的认知下降,结合其辅助检查及基因检测结果最终得到确诊。该疾病初始症状较轻,且进展缓慢[23],误诊率高,目前在国内确诊病例仅十余例,其无明显的身体缺陷,预期寿命不受影响[24],但肌阵挛发作及视力下降等症状随着时间的推移逐渐恶化,将影响患者生活质量,严重的肌阵挛使患者依赖轮椅,因此早期诊断并给予积极治疗,可以有效减轻患者痛苦,也为未来发现更精确的治疗方法提供研究基础。
对于唾液酸沉积症患者来说,对症治疗为主要治疗方法,传统的一线药物主要为丙戊酸钠,其次有氯硝西泮和苯巴比妥等[25]。本文2例患者使用抗癫痫药后未再出现强直阵挛发作,但其肌阵挛及视物模糊症状仍逐渐加重。可以推断,强直阵挛发作容易被抗癫痫药物控制,但针对肌阵挛及视力下降等的治疗仍具有挑战性,这也是目前治疗该疾病面临的主要难题。目前临床上用于指导治疗和疗效评估的指标较少,主要包括脑电图、诱发电位等神经电生理检查[24],以及通过癫痫发作频率和严重程度、日常生活功能评分进行评估[26],此外,也可使用MMS和UMRS量表评估肌阵挛严重程度[27]。
除了对症治疗,目前针对唾液酸沉积症基础病因的治疗尚处于起步阶段,其中包括酶替代疗法[28],但研究发现重组的NEU1酶会导致严重的过敏反应,因此该疗法暂时无法应用[29];除此之外,保护性蛋白/组织蛋白酶(protective protein/cathepsia A,PPCA)伴侣介导的基因治疗在小鼠模型中被证实是成功的[30-32],部分Ⅰ型患者可通过增加PPCA伴侣蛋白的水平间接增加NEU1活性[32],这在未来可能是有效的治疗方法。
