一个表皮松解性掌跖角化症家系的KRT9基因突变研究
2023-11-13姚勇丰骆志杰张泽乔阳芳
姚勇丰, 骆志杰, 张泽乔, 阳芳
1.深圳市人民医院,暨南大学第二临床医学院,南方科技大学第一附属医院, 广东 深圳 518020; 2.国家皮肤与免疫疾病临床医学研究中心分中心建设单位, 广东 深圳 518020; 3.南方科技大学医学院, 广东 深圳 518055; 4.南方医科大学皮肤病医院, 广东 广州 510091
表皮松解性掌跖角化症(epidermolytic palmoplantar keratoderma,EPPK)是掌跖角化症中最常见的类型之一,为高度遗传异质性的常染色体显性遗传病[1]。全球发病率为1.0/100 000~4.4/100 000,患者的子女有50%的概率患病,且发病无性别差异。其临床特征是弥漫性的皮肤角化过度,只局限于手掌和跖部,常有清晰的红斑边界,可伴随指挛缩、指屈曲畸形、缺指、指节垫、多汗症等症状[2]。在组织病理学上,可见表皮角化过度,棘层和颗粒层空泡变性和细胞溶解[3]。本研究对临床中收集到的一个EPPK家系展开研究,以丰富EPPK病例,并为该患者提供遗传咨询的基础。
1 资料与方法
1.1 临床资料
先证者女, 17岁,出生后数月手掌和足跖部位的皮肤出现小的角化过度斑块,逐渐扩大,并出现清晰的红斑边界,现为弥漫性角化过度斑块,呈淡黄色,表面光滑,同时伴有皲裂、脱屑,夏季减轻,冬春季加重,足底斑块偶会疼痛而影响运动。体格检查:一般情况良好,无黏膜、牙齿等异常,各系统无异常。皮肤科检查:手掌及足跖可见弥漫性角化过度斑块,质硬,呈淡黄色,光滑,界清,手掌脱屑,足跖有皲裂和脱屑(图1A、1B)。实验室检查:血常规、尿常规、粪便常规及肝肾功能检查未见明显异常,足底真菌检查为阴性。临床考虑为表皮松解性掌跖角化症。

图1 先证者临床图片 1A:先证者手掌皮肤弥漫性角化过度增厚,无指屈曲畸形和指节垫;1B:足跖皮肤弥漫性角化过度增厚,呈黄色,有皲裂和干燥鳞屑Figure 1 Clinical pictures of the proband. 1A:Diffuse hyperkeratosis on the palm,no flexion deformity of the figures and knuckle pads; 1B: Diffuse yellow hyperkeratosis on the feet, skin fissures and dry scales.
1.2 方法
1.2.1 外周血采集及DNA提取 研究经医院伦理委员会审批通过(批件号LL-KY-2023156-01)。向患者及其父母充分阐明本研究目的及意义,获取知情同意后,使用EDTA抗凝管采集患者及其父母血样各4 mL,采用QIAGEN试剂盒按说明书方法提取基因组DNA。
1.2.2 全外显子组高通量测序 简要实验流程及方法如下:①将提取的基因组DNA采用紫外分光光度仪进行纯度及浓度检测,符合全外显子组高通量测序质检要求后行下一步;②将基因组DNA打断后制备文库。外显子捕获,经PCR线性扩增后进行文库质检,合格即可利用Illumina HiSeq novas 6000测序平台测序;③对检测到的变异进行注释,注释数据库主要包括人口数据库,如gnomAD (http://gnomad.broadinstitute.org/)、1000基因组计划(http://browser.1000 genomes.org)、Berrybig data人口数据库、dbSNP (http://www.ncbi.nlm.nih.gov/snp)等;预测基因致病性的数据库,如SIFT (http://sift.jcvi.org)、FATHMM (http://fathmm.biocompute.org.uk)、MutationAssessor (http://mutationassessor.org)、CADD (http://cadd.gs.washington.edu)、SPIDEX(http://tools.genes.toronto.edu/)等;以及疾病和表型数据库,如OMIM (http://www.omim.org)、ClinVar (http://www.ncbi.nlm.nih.gov/clinvar)、HGMD (http://www.hgmd.org)、HPO (https://hpo.jax.org/app/)等。
1.2.3 Sanger测序验证 根据需要测序的DNA片段合成引物,用聚合酶链式反应(polymerase chain reaction,PCR)法进行扩增,用ABI3730xl测序仪(美国Applied Biosystems公司)以Sanger测序法进行测序,测序结果采用Snap Gene Viewer软件与Ensemble网站公布的DNA序列对比。
2 结果
2.1 家系调查
患者父母非近亲结婚。家系中4代16人,共有EPPK患者7例,男4例,女3例(图2),呈常染色体显性遗传。家系中患者均在出生后1年内发病,出现随着年龄增加而加重的掌跖弥漫性角化过度,局部皮损出现鳞屑、皲裂。先证者的母亲(Ⅲ5)同时伴有手指挛缩,家族中其他患者无指节垫、指挛缩、指屈曲畸形等其他症状。
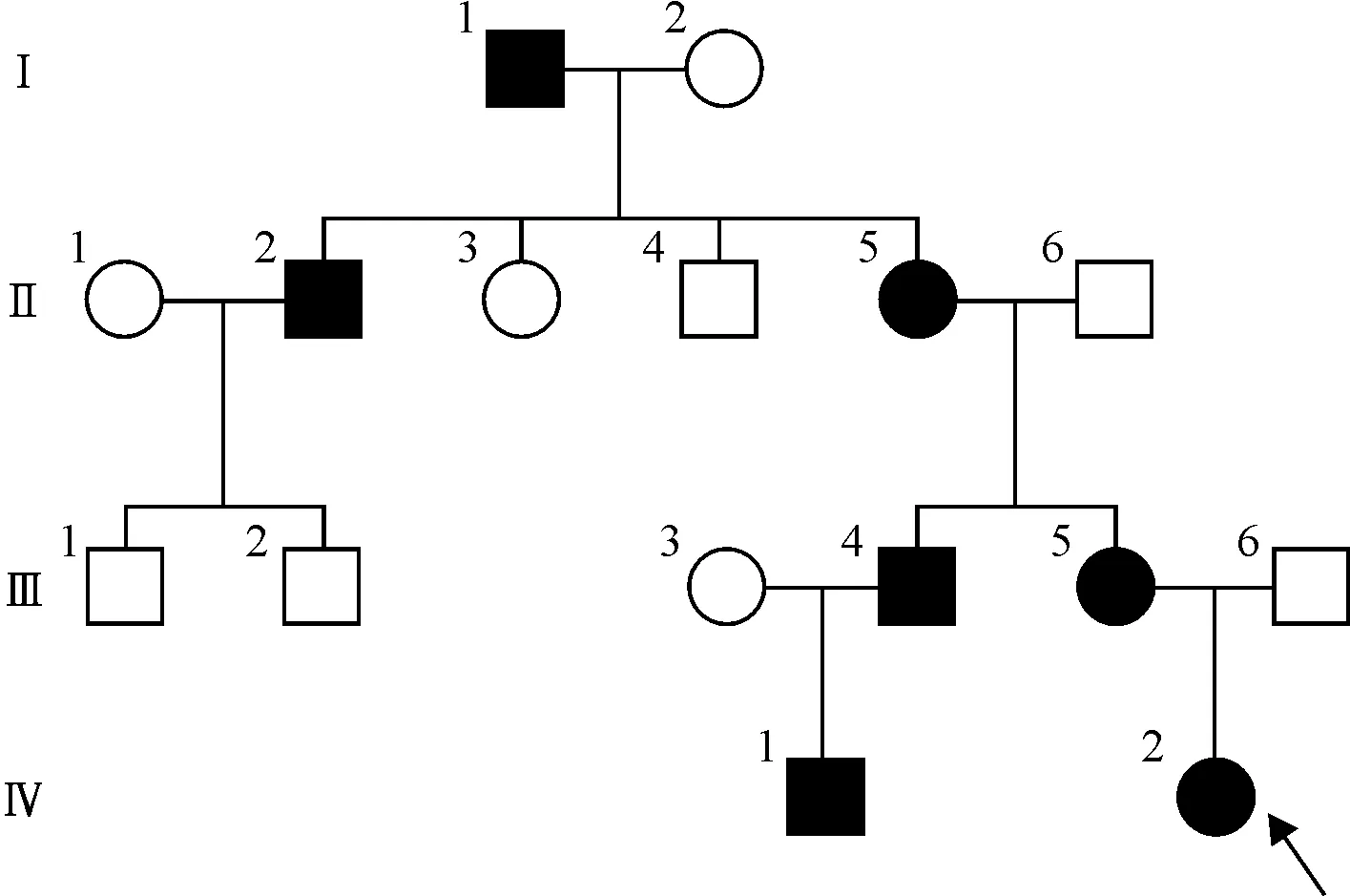
图2 表皮松解性掌跖角化症家系图(箭头示先证者)Figure 2 The family pedigree of epidermolytic palmoplantar keratoderma (The arrow indicates the proband).
2.2 WES测序结果分析和Sanger测序验证
全外显子测序发现先证者(Ⅳ-2)的基因KRT9发生c.482A>G杂合错义突变,突变导致天冬氨酸替换成丝氨酸(p.Asn161Ser),先证者的母亲(Ⅲ-5)发生该位点的杂合突变,未患病的父亲(Ⅲ-6)该位点未发生突变。Sanger测序结果与WES检测结果一致(图3)。
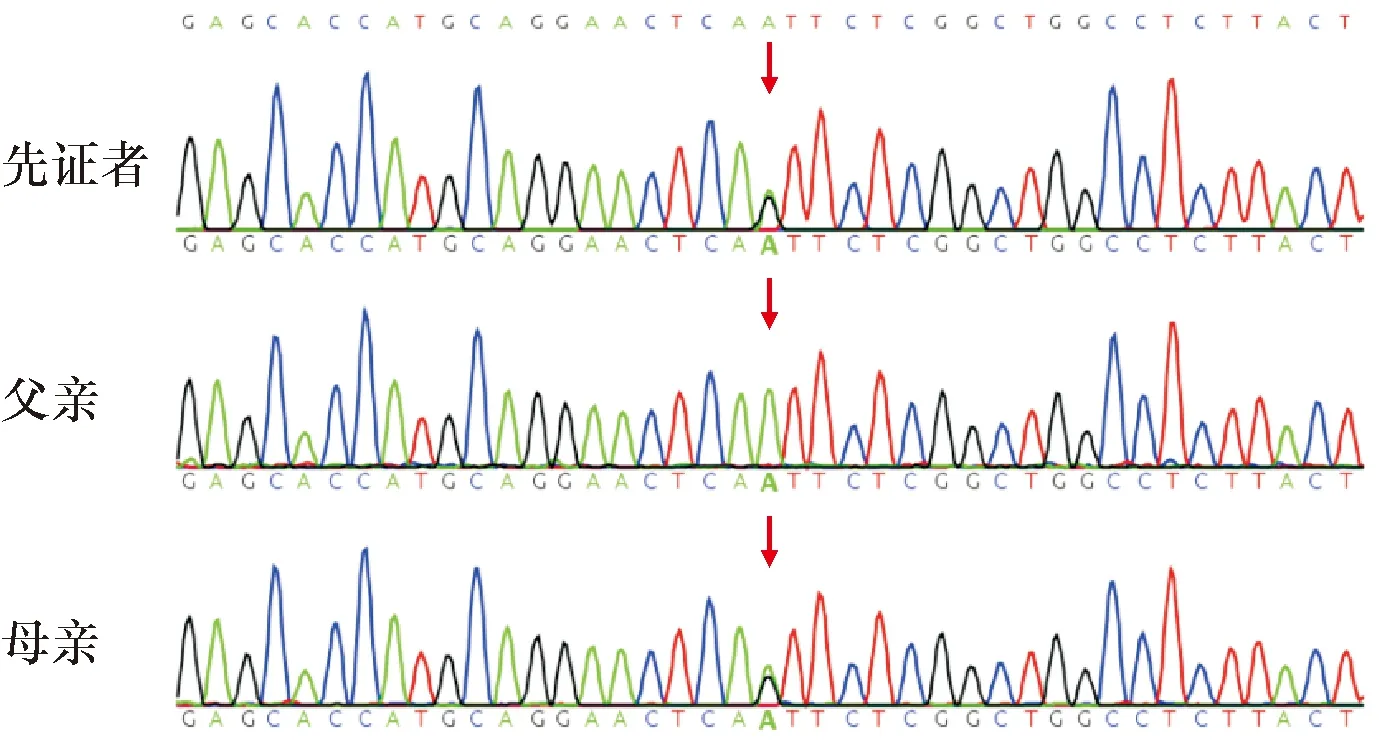
图3 Sanger 测序验证结果(箭头示突变位点)Figure 3 The results of Sanger sequencing validation (The arrows indicate the mutation site).
3 讨论
EPPK于1901年首次被Vörner发现报告,该病通常在出生时或出生后几周出现症状,仅少数于幼儿期发病。该病的临床特征初为掌跖红斑,随后出现角化过度,逐渐加重并持续终生,角化过度可延伸到手指和脚趾的侧面,常为弥漫性分布,偶为条纹状、点状、局灶分布,斑块为厚且均匀的黄色角化,但少数病例表现为深裂缝或凹坑,或出现薄的红斑边缘,极少数病例出现手背部小的角化斑块[4]。EPPK患者可以出现指节垫、与摩擦有关的皮肤损害、手掌挛缩等伴随症状,一般不累及粘膜、腺样体或牙齿等其他器官。同一突变位点可导致严重程度不同的角化和伴随症状[5]。
EPPK的主要致病基因是KRT9和KRT1,均位于17号染色体,KRT9的表达产物Ⅰ型角蛋白9是一条仅在掌跖的最终分化表皮中表达的中间丝链[6],其表达丰富使掌跖能够承受机械应力。角蛋白都由N端的头部结构域、中心的α-螺旋棒状结构域和C端尾部结构域组成,中心是由约310个氨基酸组成的α-螺旋杆状结构域,周围是非螺旋的头部(V1)和尾部(V2)结构域,杆状结构域又分为1A、1B、2A和2B段[7]。角蛋白9突变集中在1A螺旋区和2B螺旋区,即KRT9基因突变热点[8-9],突变影响中间丝网状系统的形成而损坏细胞骨架,导致角质形成细胞的脆性问题[10]。本研究所报道家系的KRT9基因第482位腺嘌呤错义突变为鸟嘌呤(c.482A>G),导致第161位天冬氨酸替换成丝氨酸(KRT9_N161S),该位点已有文献报道[2]。正常KRT9基因编码的角蛋白9通常与角蛋白1形成异二聚体,是细胞骨架中间丝的重要组成部分。Wang等[11]报道了KRT9_R163W突变导致的异常异二聚体使角质形成细胞的细胞骨架从整齐有序的正常结构变为错综复杂的网状结构,部分细胞骨架结构消失,而当用siRNA沉默KRT9_R163W时,细胞骨架恢复正常。KRT9_R163W是功能域1Aα螺旋区的第10位氨基酸,由此推测此螺旋区域是角蛋白9的功能区。距离该位点2个氨基酸的KRT9_N161S同样位于此螺旋区,可能通过抑制细胞骨架功能蛋白对体内角蛋白网络造成破坏。除KRT9外,KRT1、KRT10和KRT16的突变也可引起角蛋白结构和功能异常而导致EPPK[12]。EPPK表型严重程度取决于突变导致的角蛋白异常结构域的位置,也与表观遗传有关[4]。
KRT9编码的角蛋白9仅位于掌跖部位,但KRT1的产物角蛋白1表达于全身表皮的棘层和颗粒层细胞,KRT1突变除了导致EPPK,还引起大疱性鱼鳞病样红皮病(bullous ichthyosiform erythroderma, BCIE)、弥漫性非表皮松解性掌跖角化症(non-epidermolytic palmoplantar keratoderma, NEPPK)、表皮松解性角化过度型鱼鳞病(epidermolytic hyperkeratosis, EHK)等疾病。其已知的突变位点大多引起表型较重的BCIE,少数位点引起表型较轻的掌跖角化症(包括EPPK或NEPPK)。角蛋白1是胞浆内细胞骨架的一种Ⅱ型中间丝链,与角蛋白10结合形成卷曲的异二聚体。引起掌跖角化症的突变主要影响位于头部结构域、1A、1B 和 2B区域的编码序列,突变的角蛋白会通过扰乱角蛋白丝的组装而在细胞质内形成角蛋白团块,同时会引起细胞内其他异二聚体伴侣数量(如角蛋白10、9)的异常改变[13]。KRT1突变可引起复杂的临床异质性,同一位点突变可导致角质增厚、指节垫、甲板增厚浑浊、指趾屈曲畸形等不同的临床表现。Mo等[14]描述一家系携带c.457C>T(p.Gln153)突变,KRT1的纯合子无义突变导致先证者带有指关节垫的EPPK,而其携带杂合子突变的父母并没有显示任何临床表型,表明半剂量的角蛋白1足以维持角蛋白网络的完整性。本文报道的家系为KRT9基因杂合突变,即可引起严重的临床表现,故角蛋白9为重要的角蛋白网络组成部分。
EPPK目前除外用润肤乳和口服维生素A之外尚无特殊治疗方法。如经内科治疗无效而严重影响生活质量者,可切除掌跖过度角化的皮肤后行皮肤移植术,需切除表皮、真皮层和皮下组织,有时可根据部位深入到深筋膜,术中应注意完全切除表皮,因表皮附属物、真皮等保留了分化能力的细胞可能会导致复发[15]。基因治疗是单基因疾病治疗的最佳手段。有学者提出使用小干扰RNA(siRNA)、短发夹RNA(shRNA)等对变异基因进行CRISPR-Cas9敲除[11,16-17],以达到基因治疗EPPK的目的。该病体外实验目前亦有进展,靶向KRT9/p.Arg163Trp的siRNA在体外培养的HaCaT细胞的实验和靶向KRT9/p.Arg163Gln的siRNA在小鼠模型的实验,均证明了特异性siRNA可抑制突变KRT9在mRNA和蛋白质水平上的表达,并且小鼠的症状也有所改善[11,16]。有研究报道利用KRT9的一个插入-缺失突变体构建人类EPPK样表型的Krt9+/mut和 Krt9mut/mut小鼠模型,小鼠表现出明显的EPPK样表型。之后使用突变特异性shRNA治疗EPPK小鼠,小鼠的皮肤形态和功能恢复正常[17]。与siRNA相比,shRNA治疗具有高效力、低拷贝、持续时间长的优势,所以上调和下调的脱靶转录物比相应的siRNA更少,激活时间更长,减少了注射次数[18]。未来可能通过这类基因治疗来治疗EPPK。
综上所述,本研究详细描述了一个EPPK家系,患者由KRT9基因1号外显子发生c.482A>G(p.Asn161Ser)错义突变导致。EPPK可根据其临床表现结合基因突变分析确诊,但其致病机制仍有待进一步研究。目前动物体内有效的基因治疗为该病的临床基因治疗提供了理论基础,是非常有前景的治疗方法。
